Prepare for MRCP Examination

How to prepare for the MRCP Examination?
To prepare for part 1 of the MRCP examination, you must have a solid knowledge of internal medicine, including hematology, hemato-oncology, and genetics, along with expert insights. This article will cover the hematology and hemato-oncology topics relevant to part 1 of the UK MRCP examination.
What is the MRCP examination?
The Membership of the Royal Colleges of Physicians (MRCP) examination in the United Kingdom is a prestigious and comprehensive assessment for medical professionals, including all subspecialists in internal medicine. Developed by the Royal Colleges of Physicians of London, Edinburgh, and Glasgow, the MRCP is a key milestone in the career of physicians seeking to advance their expertise and gain recognition in internal medicine. The Royal College of Physicians of Ireland (MRCPI) examination in General Medicine is accredited by the Medical Council of Ireland as the foremost knowledge-based assessment for internal medicine in Ireland. If you are undertaking Basic Specialist Training in Ireland, achieving MRCPI means you are ready to apply to enter Higher Specialist Training, the final stage of training before independent practice.
The examination is designed to assess the clinical knowledge, skills, and competencies of physicians, ensuring they meet the highest standards of medical practice. Divided into three parts – MRCP Part 1, Part 2 Written, and Part 2 Practical Assessment of Clinical Examination Skills (PACES) – the MRCP examination covers a wide range of medical disciplines, reflecting the diverse and evolving nature of internal medicine. Successful candidates who pass all three parts are eligible to apply for the award of the MRCP(UK) Diploma.
While curating this article on “Prepare for MRCP Examination”, I stumbled upon an invaluable resource I am eager to share with fellow candidates. Dr Keith Patterson, a distinguished consultant haematologist based in the UK, has created an exceptional YouTube video on behalf of Pastest, specifically addressing the MCQ questions on haematology topics relevant to the MRCP Part 1 examination. I found these videos to be an indispensable aid during candidates’ preparation, providing clarity and depth to the subject matter. To optimize your learning experience, I highly recommend watching the video before delving into my detailed comments on each topic. This approach allows you to test your knowledge firsthand and leverage Dr. Patterson’s expertise to enhance your understanding. Combining this insightful video and my comprehensive insights will equip you with a robust foundation for success in your MRCP journey.
Sideroblastic Anemias:
Sideroblastic anemia is a type of anemia that results from abnormal utilization of iron during erythropoiesis. There are different forms of sideroblastic anemia, and all forms are defined by the presence of ring sideroblasts in the bone marrow.
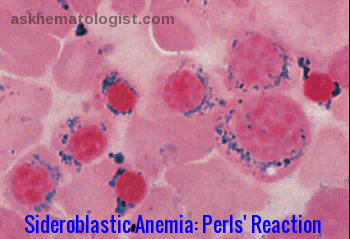
Ring sideroblasts are erythroid precursors containing deposits of non-heme iron in mitochondria, forming a ring-like distribution around the nucleus. The iron-formed ring covers at least one-third of the nucleus rim. Sideroblastic anemia is known to cause microcytic and macrocytic anemia, depending on what type of mutation led to it. Unlike iron deficiency anemia, where there is a depletion of iron stores, patients with sideroblastic anemia have normal to high iron levels. The iron-containing granules (demonstrated by Prussian blue stain – Perls’ reaction) surround the nuclei of some erythroblasts.
- The disorder may be hereditary or acquired.
- The acquired form may be secondary to certain drugs (e.g. antituberculous agents like isoniazid), lead poisoning, other toxic conditions, nutritional deficiencies (copper, vitamin B6), alcohol, as a clonal disorder (myelodysplasia) which may progress to acute leukemia or idiopathic.
- Treatment of sideroblastic anemia may include removal of toxic agents; administration of pyridoxine, thiamine, or folic acid; transfusion (along with antidotes if iron overload develops from transfusion); other medical measures; bone marrow or liver transplantation.
Read more: https://askhematologist.com/miscellaneous-red-cell-abnormalities/#ixzz8LXA6IAOx
Hemochromatosis: 
Hereditary Hemochromatosis (HH) is a genetic disorder that causes the body to absorb too much iron (Fe) from the diet. The excess iron is stored in the body’s tissues and organs, particularly the skin, heart, liver, pancreas, and joints.
Because humans cannot increase the excretion of iron, excess iron can overload and eventually damage tissues and organs. For this reason, hereditary hemochromatosis is also called an iron overload disorder.
Early symptoms of HH are nonspecific and may include fatigue, joint pain, abdominal pain, and loss of sex drive. Later signs and symptoms can include arthritis, liver disease, diabetes, heart abnormalities, and skin discoloration. 
Although penetrance is more commonly an issue with autosomal dominant disorders, it appears to be an issue in hereditary hemochromatosis. Thirty to fifty percent of those with homozygous genotypes do not have clinical evidence of hemochromatosis. Many factors, including alcohol consumption, dietary iron intake, blood loss associated with menstruation and pregnancy and blood donation, influence the expression.
Hereditary hemochromatosis is classified by type depending on the age of onset and other factors such as genetic cause and mode of inheritance.
There are 4 types of HH, types 1 through 4.
Type 1, is the most common form of the disorder, and type 4 (also called ferroportin disease) begins in adulthood. Men with type 1 or type 4 hemochromatosis typically develop symptoms between the ages of 40 and 60, and women usually develop symptoms after menopause.
Type 2 hemochromatosis is a juvenile-onset disorder. Iron accumulation begins early in life, and symptoms may appear in childhood. By age 20, decreased or absent secretion of sex hormones is evident. Females usually begin menstruation in a normal manner, but menses stop after a few years. Males may experience delayed puberty or symptoms related to a shortage of sex hormones. If the disorder is untreated, heart disease becomes evident by age 30.
The onset of type 3 hemochromatosis is usually intermediate between types 1 and 2. Symptoms of type 3 hemochromatosis generally begin before age 30.
Phlebotomy (venesection) is the simplest and most effective method to remove excess iron. It delays the progression of fibrosis to cirrhosis, sometimes even reversing cirrhotic changes, and prolongs survival, but it does not prevent hepatocellular carcinoma.
About 500 mL of blood (about 250 mg of iron) is removed weekly or biweekly (every other week) until serum ferritin levels reach 20 to 50 ng/mL. Weekly or biweekly phlebotomy may be needed for many months (e.g. if 250 mg of iron are removed per week, 40 weeks will be required to remove 10 g of iron). When iron levels are normal, phlebotomies can be intermittent to maintain ferritin between 50 and 100 ng/mL. 
Read more: https://askhematologist.com/hemochromatosis/#ixzz8LXImC9sP
Sickle Cell Anaemia: 
Sickle cell disease (HbS) is a severe hereditary form of anemia in which a mutated form of hemoglobin distorts the red blood cells (RBC’s) into a crescent shape at low oxygen levels.
Sickle cell disease (HbS) is common among those of African descent.
In this disease, a single base mutation in the β-globin gene leads to the substitution of valine for glutamine at the 6th amino acid position of the β-globin chain. High levels of deoxygenated sickle Hb form reversible fibrils leading to sickling of the red cells.
The abnormal Hb SS is prone to form tactoids with crystallization in the RBC’s when oxygen tension is low, and the RBC’s change shape to long, thin sickle cells that are “sticky” and sludge in capillaries, further decreasing blood flow and oxygen tension. The sickled RBCs tend to adhere to endothelium, and the bioavailability of endothelial nitric oxide is reduced as well, further promoting vaso-occlusion. 
 Read more: https://askhematologist.com/sickle-cell-disease/#ixzz8LXN40qZv
Read more: https://askhematologist.com/sickle-cell-disease/#ixzz8LXN40qZv
Thrombocytosis:
Platelets are parts of the blood that help form blood clots. Thrombocytosis is a disorder in which the body produces too many platelets.
Platelets are acute-phase reactants; therefore, they increase in response to various stimuli, including systemic infections, inflammatory conditions, bleeding, and tumors.
It’s called reactive thrombocytosis or secondary thrombocytosis when the cause is an underlying condition, such as an infection.
Less commonly, when the high platelet count has no apparent underlying condition as a cause, the disorder is called primary thrombocythemia or essential thrombocythemia. This is a blood and bone marrow disease.

Essential (1ry) Thrombocythemia
A high platelet level may be detected in a routine blood test. It’s important to determine whether it’s reactive thrombocytosis or essential thrombocythemia to choose the best treatment options.
Essential thrombocythemia (ET) is a myeloproliferative neoplasm (MPN) characterized by megakaryocyte hyperplasia and thrombocytosis due to a clonal abnormality of a multipotent hematopoietic stem cell.
From the genetic perspective, ET patients harbour mutations in JAK2 (50–60%), CALR (15–30%) and MPL (1–5%) genes.
Platelet survival is normal in ET.
Approximately one-third of patients with ET are asymptomatic at diagnosis.
 Read more: https://askhematologist.com/essential-thrombocythemia/#ixzz8LXcaCLUG
Read more: https://askhematologist.com/essential-thrombocythemia/#ixzz8LXcaCLUG
Megaloblastic Anemias:
Megaloblastic anemias are a group of disorders characterized by reduced DNA synthesis (not just in red cell series) associated with pathognomonic morphological changes. When DNA synthesis is impaired, the cell cycle cannot progress from the G2 growth stage to the mitosis (M) stage. This leads to continuing cell growth without division, which presents as macrocytosis. In other words, megaloblastic anemia is a condition in which the bone marrow produces unusually large, structurally abnormal, immature red blood cells (megaloblasts).

Macrocytosis is a term used to describe erythrocytes that are larger than normal, typically reported as mean cell volume (MCV) greater than 100 fL. The amount of hemoglobin increases proportionately with the increase in cell size. Therefore, if the increase in MCV is not related to macrocytic anemia, the mean cell hemoglobin concentration (MCHC) also increases in proportion.
Causes of macrocytosis are many and range from benign to malignant; thus, a complete workup to determine etiology is essential. Macrocytosis may occur at any age, but it is more prevalent in older age groups because the causes of macrocytosis are more prevalent in older persons.
The defect in red cell DNA synthesis is most often due to hypovitaminosis, specifically a deficiency of vitamin B12 and/or folic acid. 

Blood smear showing hypersegmented neutrophil
The pathological state of megaloblastosis is characterized by many large immature and dysfunctional red blood cells (megaloblasts) in the bone marrow and also by hypersegmented neutrophils (those exhibiting five or more nuclear lobes “segments”). These hypersegmented neutrophils are found in the “peripheral blood”.
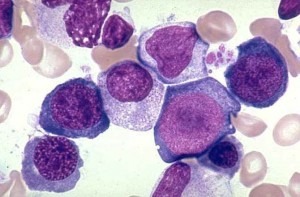
Megaloblasts in Bone Marrow
Read more: https://askhematologist.com/megaloblastic-anemias/#ixzz8Lc18s9yd
Microangiopathic Hemolytic Anemia:
Microangiopathic hemolytic anemia (MAHA) is now used to designate any hemolytic anemia related to RBC fragmentation, occurring in association with small vessel disease. In disseminated intravascular coagulation (DIC), RBC fragmentation is thought to result from the deposition of fibrin or platelets within the microvasculature. The term “thrombotic microangiopathy (TMA)” is also used to describe syndromes characterized by MAHA, thrombocytopenia, and thrombotic lesions in small blood vessels. The most prominent diagnoses associated with TMA are thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS). Many different disorders, including preeclampsia, infections, adverse drug reactions, hematopoietic stem cell transplantation, autoimmune diseases, and malignancies, can cause TMA (i.e., secondary TMA). Recently, because the pathogeneses of TTP and HUS have been elucidated, great progress has been made in diagnosis and treatments. However, the pathogenesis of secondary TMA remains unclear.
In thrombotic microangiopathies and thrombotic thrombocytopenic purpura a pentad of signs is classically described—thrombocytopenia (platelet counts usually less than 20,000/mm3), microangiopathic hemolytic anemia, fever, renal dysfunction, and neurologic signs. A clinical triad of thrombocytopenia, red blood cell fragments (schistocytosis), and an increased lactate dehydrogenase (LDH) level is enough to suggest the diagnosis. Examination of the peripheral blood smear in patients with thrombocytopenia of unclear cause is imperative to exclude this diagnosis.

Peripheral blood smear of a patient with microangiopathic hemolytic anemia showing schistocytes (arrows).
Acute DIC manifests with bleeding and oozing from multiple sites, catheter access or mucosal surfaces, often in a critically ill patient with multisystem organ failure. Chronic DIC is most often associated with malignancy, usually solid tumors, and results from continuous slow exposure of blood to small amounts of tissue factor without overwhelming the compensatory mechanisms that regenerate depleted factors. It most often manifests clinically with thrombosis rather than hemorrhage.
The diagnosis of acute DIC relies on the following: history and clinical setting, with particular attention to trauma, sepsis, malignancy, pregnancy, and miscarriages; moderate to severe thrombocytopenia; evidence of microangiopathic hemolysis on the peripheral smear (e.g., the presence of schistocytes); and suggestive laboratory test results. Clinically significant DIC is unlikely in the presence of normal d-dimers. Prolonged PT and aPTT can also be noted, as well as decreased fibrinogen levels; fibrinogen, however, is an acute-phase reactant and may be falsely normal. The thrombin time (TT) is prolonged, whereas antithrombin III (ATIII), protein C, and protein S levels are often depressed.
Chronic DIC may manifest with more subtle laboratory results—smear microangiopathy and an elevated d-dimer level may be the only laboratory finding.
Read more: https://askhematologist.com/thrombocytopenia/#ixzz8LccKtMJi
Malaria:
Malaria is due to infection with specific protozoa of the Plasmodium genus. It is transmitted by the bite of an infective female Anopheles mosquito. The malaria parasite undergoes a single sexual cycle in the mosquito and recurrent asexual cycles, with the production of sexual forms (gametocytes) in man.
Clinical features:
- The initial incubation period of malaria is 9-11 days.
- Malaria symptoms include fever, shaking chills, body aches, cough, headache, anemia, fatigue, malaise, and splenomegaly. The fever is often periodic, the frequency in part reflecting the species of Plasmodium.
- In severe cases especially of the malignant tertian form (P. falciparum), there may be hemolysis, thromboses, shock, and cerebral complications (Blackwater Fever). Blackwater fever is a severe clinical syndrome occurring as a complication of malarial infection characterized by intravascular hemolysis, hemoglobinuria, and acute renal failure in people exposed to Plasmodium falciparum and, to some extent, in people who were exposed to medications like quinine and mefloquine.
It was demonstrated in the late eighties and early nineties that those with O type blood group are protected from severe or fatal Plasmodium falciparum malaria compared with non-O blood groups (A, B, and AB) by the mechanism of inducing high levels of anti-malarial IgG antibodies.
Investigations:
During an acute episode of malaria, there may be anemia and plasmodial forms can be detected in the peripheral blood, especially on thick and thin blood films. Differentiation of the different species requires considerable expertise but is important as together with the geographical source of origin, this will influence the choice of therapy. Note that 1 negative smear does not exclude malaria as a diagnosis; several more smears should be examined over 36 hours.
Specific tests for malaria infection should be also carried out e.g. Microhematocrit centrifugation and Fluorescent dyes/ultraviolet indicator tests.
Fewer than 5% of malaria patients have leucocytosis; thus, if leucocytosis is present, the differential diagnosis should be broadened!
If the patient is to be treated with primaquine, glucose-6-phosphate dehydrogenase (G6PD) level should be checked.
Plasmodium Falciparum:

Peripheral blood smear showing trophozoites in various stages of development with signet-ring classically seen in Plasmodium falciparum infection.

Parasites may be scanty, but red cells super-infected with trophozoites may be seen. Infected RBCs are round, of normal size and Schuffner’s dots are not seen. The gametocytes are typically elongated or banana-shaped.
Plasmodium Vivax:

Plasmodium Vivax Trophozoites
Ring forms (trophozoites) and segmented schizonts may be seen. The schizonts may develop into free merozoites or into round sexual forms (gametocytes).
The invaded red cells are increased in size and contain Schuffner’s dots.
Plasmodium Ovale:
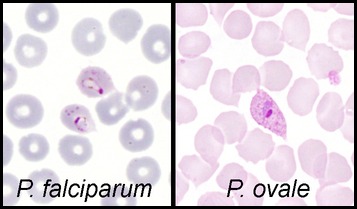
The appearances in the blood are similar to Plasmodium Vivax except that the gametocytes and infected red cells are often oval with fimbriated edges. Schuffner’s dots are conspicuous.
Plasmodium Malariae:
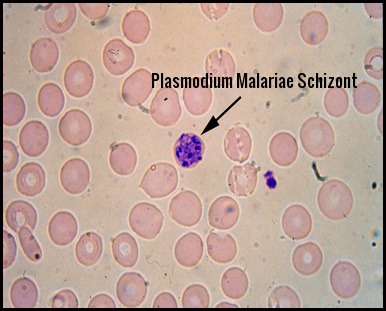
In this form, the trophozoites are often band-shaped. The infested red cells are not enlarged and there are no Schuffner’s dots.
N.B. It should be noted that mixed infections can occur.
Prophylaxis:
Blood type, metabolism, exercise, shirt color and even drinking beer can make individuals especially delicious to mosquitoes! Mosquitoes bite us to harvest proteins from our blood—research shows that they find certain blood types more appetizing than others. One study found that in a controlled setting, mosquitoes landed on people with Type O blood nearly twice as often as those with Type A. People with Type B blood fell somewhere in the middle of this itchy spectrum. Additionally, based on other genes, about 85 percent of people secrete a chemical signal through their skin that indicates which blood type they have, while 15 percent do not, and mosquitoes are also more attracted to secretors than nonsecretors regardless of which type they are.
Prophylaxis involves mosquito control, avoidance of bites and appropriate prophylactic drug therapy. Avoid mosquitoes by limiting exposure during times of typical blood meals (ie, dawn, dusk). Wearing long-sleeved clothing and using insect repellants may also prevent infection. Avoid wearing perfumes and colognes.
Considerations when choosing a drug for malaria prophylaxis:
- Recommendations for drugs to prevent malaria differ by country of travel and can be found in the country-specific tables of the Yellow Book. Recommended drugs for each country are listed in alphabetical order and have comparable efficacy in that country.
- No antimalarial drug is 100% protective and must be combined with the use of personal protective measures, (i.e., insect repellent, long sleeves, long pants, sleeping in a mosquito-free setting or using an insecticide-treated bednet).
- For all medicines, also consider the possibility of drug-drug interactions with other medicines that the person might be taking as well as other medical contraindications, such as drug allergies.
Treatment:
Consider malaria in every febrile patient returning from a malaria-endemic area within the last year, especially in the previous three months, regardless of whether they have taken chemoprophylaxis, as prompt recognition and appropriate treatment will improve prognosis and prevent deaths. Failure to consider malaria in the differential diagnosis of febrile illness in a patient who has travelled to an area where malaria is endemic can result in significant morbidity or mortality, especially in children and in pregnant or immunocompromised patients.
Malaria treatment:
- Rest and fluids are required during the acute phase.
- Drugs are given to eradicate the asexual blood-borne cycle and the exoerythrocytic (liver) parasites “don’t occur with P. falciparum”.
- Several groups of drugs are available and the correct drug should be chosen by an expert.
- Generally speaking, P. falciparum is resistant to Chloroquine treatment. Resistance is rare in P. vivax infection, and P. ovale and P. malariae remain sensitive to chloroquine. Primaquine is required in the treatment of P. ovale and P. vivax infections to eliminate the exoerythrocytic (liver) parasites.
Read more: https://askhematologist.com/malaria/#ixzz8LdF5QxsB
Aplastic Anemia:
Aplastic anemia (AA) is a rare disease in which the bone marrow and the hematopoietic stem cells that reside there are damaged. This causes a deficiency of all three blood cell types (pancytopenia): red blood cells (anemia), white blood cells (leucopenia), and platelets (thrombocytopenia). Aplastic refers to the inability of the stem cells to generate mature blood cells.
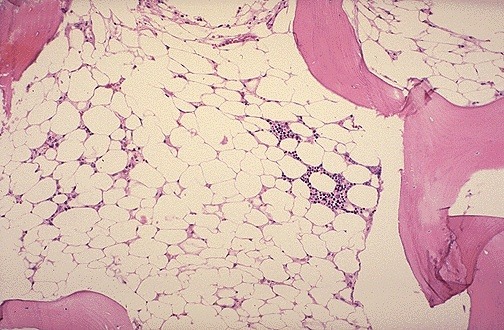
Severe hypoplastic marrow in aplastic anemia
Aetiology:
Congenital:
The commonest of these disorders is Fanconi’s anemia. This is an autosomal recessive disease usually presenting at 4-7 years. Bony and renal abnormalities are common. The erythrocyte sedimentation rate (ESR) is usually high, the bone marrow appears megaloblastic and HbF is often high.
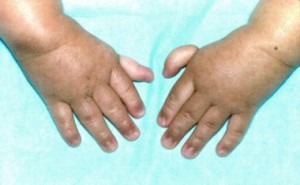
A 3-year-old patient with Fanconi anemia (rudimentary thumbs)
Acquired:
Acute:
Usually due to infections e.g. parvovirus B19 and acute viral hepatitis infection but can be caused by any of the acquired causes.
Chronic:
- Idiopathic (about half the cases).
- Drugs (e.g. chloramphenicol, carbamazepine, felbamate, phenytoin, quinine, and phenylbutazone), Chemicals (e.g. benzene), irradiation and chemotherapy.
- Infections especially viral hepatitis. Also, Epstein-Barr virus, cytomegalovirus, parvovirus B19, and HIV.
- An autoimmune disorder in which white blood cells attack the bone marrow.
- Associated with paroxysmal nocturnal hemoglobinuria.
- Could rarely associate pregnancy. Sometimes, this type of aplastic anemia improves on its own after the woman gives birth.
Clinical features:
The clinical presentation of patients with aplastic anemia includes symptoms related to the decrease in bone marrow production of hematopoietic cells. The onset is insidious, and the initial symptom is frequently related to anemia or bleeding, although fever or infections may be noted at presentation.
Signs and symptoms of aplastic anemia may include the following:
- Pallor
- Headache
- Palpitations, dyspnea
- Fatigue
- Foot swelling
- Gingival bleeding, petechial rashes
- Overt and/or recurrent infections
- Oropharyngeal ulcerations
- A subset of patients with aplastic anemia present with jaundice and evidence of clinical hepatitis.
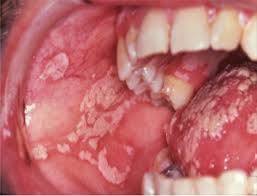
Severe oral candidiasis (thrush) in aplastic anemia
Investigations:
- Anemia (normocytic or mildly macrocytic).
- Absolute reticulocytopenia.
- Absolute granulocytopenia.
- Monocytopenia is usual.
- Thrombocytopenia.
- Bone marrow shows abundant fat spaces and few hematopoietic cells.

Bone Marrow Biopsy-Low power-H&E: hypocellular bone marrow with increased adipose tissue and decreased hematopoietic cells in the marrow space.
The condition needs to be differentiated from pure red cell aplasia. In aplastic anemia, the patient has pancytopenia (i.e., anemia, neutropenia, and thrombocytopenia). In contrast, pure red cell aplasia is characterized by a reduction in red cells only. The diagnosis can only be confirmed on a bone marrow examination. Before this procedure is undertaken, a patient will generally have had other blood tests to find diagnostic clues, including an FBC, blood film, U&Es, liver enzymes, thyroid function tests, vitamin B12 and folate levels.
The following tests aid in determining the differential diagnosis of aplastic anemia:
- Bone marrow aspirate and biopsy: to rule out other causes of pancytopenia (i.e. neoplastic infiltration or significant myelofibrosis).
- History of iatrogenic exposure to cytotoxic chemotherapy: can cause transient bone marrow suppression.
- X-rays, computed tomography (CT) scans, or ultrasound imaging tests: enlarged lymph nodes (a sign of lymphoma), kidneys and bones in arms and hands (abnormal in Fanconi anemia).
- Chest X-ray: infections.
- Liver tests: liver diseases.
- Viral studies: viral infections.
- Vitamin B12 and folate levels: vitamin deficiency.
- Blood tests e.g. flow cytometry for paroxysmal nocturnal hemoglobinuria.
- Test for antibodies: immune competency.
Treatment:
Supportive care:
- Infection is the most likely cause of death in aplastic anemia and must be treated early and aggressively. Multiple swabs and cultures should be sent before starting broad-spectrum antibiotics. Antifungal agents should be considered if the infection is not controlled with antibiotics alone e.g. Fluconazole, Voriconazole, Caspofungin. Prophylactic antiseptic mouthwash e.g. Corsodyl or Difflam mouth rinse and Mycostatin (Nystatin) mouth drops are usually prescribed.
- Red cell and platelet support must be given when required.
Active treatment:
- Treating immune-mediated aplastic anemia involves suppression of the immune system, an effect achieved by daily medicine intake, or, in more severe cases, a bone marrow transplant, a potential cure. The transplanted bone marrow replaces the failing bone marrow cells with new ones from a matching donor. The multipotent stem cells in the bone marrow reconstitute all three blood cell lines, giving the patient a new immune system, red blood cells, and platelets. However, besides the risk of graft failure, there is also a risk that the newly created white blood cells may attack the rest of the body (“graft-versus-host disease”). In young patients with an HLA-matched sibling donor, bone marrow transplant can be considered as a first-line treatment, patients lacking a matched sibling donor typically pursue immunosuppression as a first-line treatment, and matched unrelated donor transplants are considered second-line therapy.
- Medical therapy for aplastic anemia often includes a course of anti-thymocyte globulin (ATG) and several months of treatment with cyclosporin to modulate the immune system. Chemotherapy with agents such as cyclophosphamide may also be effective but has more toxicity than ATG. Antibody therapy, such as ATG, targets T-cells, which are believed to attack the bone marrow. Corticosteroids are generally ineffective though they are used to ameliorate serum sickness caused by ATG. Normally, success is judged by bone marrow biopsy 6 months after initial treatment with ATG.
- One prospective study involving cyclophosphamide was terminated early due to a high incidence of mortality, due to severe infections as a result of prolonged neutropenia.
- The milder disease can resolve with supportive care and stimulating the bone marrow with hematopoietic growth factors such as G-CSF e.g. Filgrastim (Neupogen) or Lenograstim (Granocyte) which may increase the neutrophil count.
Read more: https://askhematologist.com/aplastic-anemia/#ixzz8LhC5xLEN
Von Willebrand Disease:
Von Willebrand disease (VWD) is a blood disorder in which the blood does not clot properly. Blood contains many proteins that help the blood clot when needed. One of these proteins is called von Willebrand factor (VWF). People with VWD either have a low level of VWF in their blood or the VWF protein doesn’t work the way it should.
Normally, when a person is injured and starts to bleed, the VWF in the blood attaches to small blood cells called platelets. This helps the platelets stick together, like glue, to form a clot at the site of injury and stop the bleeding. When a person has VWD, because the VWF doesn’t work the way it should, the clot might take longer to form or form incorrectly and bleeding might take longer to stop. This can lead to heavy, hard-to-stop bleeding. Although rare, the bleeding can be serious enough to damage joints or internal organs, or even be life-threatening. 
Who is Affected?
VWD is the most common bleeding disorder, found in up to 1% of the U.S. population. This means that 3.2 million (or about 1 in every 100) people in the United States have the disease. Although VWD occurs among men and women equally, women are more likely to notice the symptoms because of heavy or abnormal bleeding during their menstrual periods and after childbirth. There are 3 major types of VWD: Type 1, Type 2, and Type 3.
Types of VWD:
Type 1 is the most common and mildest form of VWD, in which a person has lower-than-normal levels of VWF. A person with Type 1 VWD also might have low levels of factor VIII (8), another type of blood-clotting protein. About 85% of people treated for VWD have Type 1.
In type 2 although the body makes normal amounts of the VWF, the factor does not work the way it should. Type 2 is further broken down into four subtypes―2A, 2B, 2M, and 2N―depending on the specific problem with the person’s VWF. Because the treatment is different for each type, a person must know which subtype he or she has.
Type 3 is the most severe form of VWD, in which a person has very little or no VWF and low levels of factor VIII. This is the rarest type of VWD. Only 3% of people with VWD have Type 3.
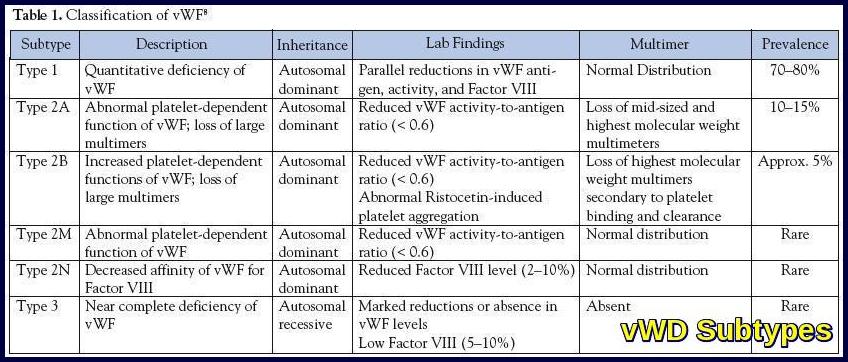
Causes:
Most people who have VWD are born with it. It almost always is inherited, or passed down, from a parent to a child. VWD can be passed down from either the mother or the father, or both, to the child.
While rare, a person can get VWD without a family history of the disease. This can happen if a spontaneous mutation occurs. That means there has been a change in the person’s gene. Whether a child receives the affected gene from a parent or as a result of a mutation, once the child has it, the child can later pass it along to his or her children.
Also, it is rare, but possible, for a person to get or acquire VWD (they didn’t receive the affected gene from their parent or as a result of a mutation) later in life because of an underlying medical condition. This can happen when a person’s immune system destroys his or her VWF, often as a result of the use of a medication or as a result of another disease. If VWD is acquired in this way, it cannot be passed along to any children.
Signs and Symptoms:
- Frequent or Hard-to-Stop Nosebleeds.
- Easy Bruising.
- Heavy Menstrual Bleeding.
- Longer than Normal Bleeding After Injury, Surgery, Childbirth, or Dental Work.

Diagnosis:
To find out if a person has VWD, the doctor will ask questions about personal and family histories of bleeding. The doctor also will check for unusual bruising or other signs of recent bleeding and order some blood tests to measure how the blood clots. The tests will provide information about the amount of clotting proteins present in the blood and if the clotting proteins are working properly. Because certain medications can cause bleeding, even among people without a bleeding disorder, the doctor will ask about recent or routine medications taken that could cause bleeding or make bleeding symptoms worse.
A combination of blood tests are used to diagnose VWD, including a VWF antigen test, which measures the amount of VWF in the blood, tests that measure clotting time and ability to form a clot, and tests measuring platelet function. Some of these tests may have to be repeated because the levels of VWF can change due to stress, exercise, the use of birth control pills, pregnancy, and hyperthyroidism. People with VWD usually have less than 50% of normal VWF in their blood. After a diagnosis of VWD is discovered, an additional is given to determine the type. The best place for patients with bleeding disorders to be diagnosed and treated is at one of the hemophilia treatment centres.
1- Complete Blood Count (CBC).
2- Activated Partial Thromboplastin Time (APTT) Test.
3- Prothrombin Time (PT) Test.
4- Fibrinogen Test.
5- Factor VIII clotting activity―To measure the amount of factor VIII in the blood.
6- Von Willebrand factor antigen―To measure the amount of VWF in the blood.
7- Ristocetin cofactor or other VWF activity―To measure how well the VWF works.
8- Von Willebrand factor multimers―To measure the makeup or structure of the VWF.
9- Platelet aggregation tests―To measure how well the platelets are working.
Bleeding time testing is unreliable and no longer done.
Treatment:
Treatment of vWD is difficult to monitor because of the lack of laboratory tests that correlate with bleeding. Hence, commonly monitored parameters include clinical bleeding, factor VIII levels, and ristocetin cofactor levels.
Desmopressin:
Desmopressin (DDAVP) promotes the release of vWF from endothelial cells. It is effective for patients with type 1 disease but has a varying effect for patients with type 2A disease. It is relatively contraindicated in patients with type 2B disease. It may also be helpful for patients with type 2M or 2N vWD but is not helpful for patients with type 3 disease. Desmopressin can be given intravenously or subcutaneously at 0.2 μg/kg (maximum dose, 20 mg), with a response noted as early as 30 minutes later, lasting 6 to 12 hours. The dose may be repeated in 12 hours and then daily. The intranasal preparation is given at a dose of 150 mg for patients weighing less than 50 kg and 300 mg for those weighing more than 50 kg. A trial infusion is needed to assess the efficacy of treatment and adequacy of prophylactic use. Adverse effects include facial flushing, headaches, hyponatremia with continuous use, and a potential for thrombotic events.
Factor VIII Concentrates:
Intermediate purity factor VIII concentrates are used for patients who do not benefit from desmopressin and for those with serious bleeding or before major surgery. Intermediate-purity factor VIII concentrate is used to maintain factor VIII levels between 50% and 100% for 3 to 10 days. A dose of 20 to 30 IU/kg is typically used twice daily. Overzealous treatment results in high factor VIII levels, which is believed to increase the risk of thrombosis.
Aminocaproic Acid and Tranexamic Acid:
Aminocaproic acid, 50 mg/kg four times daily, and tranexamic acid, 25 mg/kg three times daily, have been used for mild bleeding episodes and dental procedures. They carry a risk of thrombotic events, which is especially pronounced in older individuals and with long-term use.
Topical Treatment:
Topical treatment for oral or nasal bleeding with Gelfoam or Surgicel soaked with thrombin has been used successfully. Topical therapy plays an important role in prophylaxis and treatment after dental procedures.
Recombinant Factor VIIa:
Recombinant factor VIIa has been used successfully in patients with type 3 vWD with alloantibodies. In addition, its use should be considered in patients with life-threatening bleeding in whom other measures have failed. Disadvantages include its high cost as well as increased risks of thrombotic events, which are more pronounced in older adults.
Read more: https://askhematologist.com/qualitative-platelet-disorders/#ixzz8MCC170Mz
Exam dates and fees:

MRCP Examination Part 1 Dates and Fees – 2024
Read more: https://www.mrcpuk.org/mrcpuk-examinations/part-1/exam-dates-and-fees
References:
The Royal College of Physicians of London (RCP London). https://www.rcplondon.ac.uk/
The Royal College of Physicians of Ireland (RCP Ireland). https://www.rcpi.ie/Learn-and-Develop/Examinations/RCPI-Examinations/MRCPI-in-General-Medicine/Exam-Format/General-Medicine-Part-I
Dr Keith Patterson. Consultant haematologist, London, UK. MRCP MCQs – Haematology (Part 1).
Get the Pastest Advantage. Work through the largest bank of MRCP Part 1 questions on the market and Past Papers from recent exams https://www.pastest.com/mrcp-part-1/
Knowledge Pro@ExpertMedicalEducation‧ Channel for all MRCP Preparation Individuals.
May A, et al. Sideroblastic anemia: diagnosis and management. Hematology Am Soc Hematol Educ Program. 2017;2017(1):272-276. doi: 10.1182/ash education-2017.1.272
Ashorobi D, Chhabra A. Sideroblastic Anemia. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538287/
Pietrangelo A. Hereditary hemochromatosis: Pathogenesis, diagnosis, and treatment. Gastroenterology 139:393–408, 2010.
James Peter, Adam Hamilton. Hereditary Hemochromatosis – Hematology and Oncology – Merck Manuals Professional Edition http://www.merckmanuals.com/professional/hematology-and-oncology/iron-overload/hereditary-hemochromatosis. Last accessed January 2017.
Genetics Home Reference: hereditary hemochromatosis. https://ghr.nlm.nih.gov/condition/hereditary-hemochromatosis#
Wang WC (2009). Sickle cell anemia and other sickling syndromes. In JP Greer et al., eds., Wintrobe’s Clinical Hematology, 12th ed., pp. 1038-1082. Philadelphia: Lippincott Williams and Wilkins.
Yawn BP, et al. (2014). Management of sickle cell disease: Summary of the 2014 evidence-based report by expert panel members. JAMA, 312(10): 1033-1048.
Dr. Eric Strong, Dr. Gerald Diaz. Etiologies of thrombocytosis. https://www.grepmed.com/images/11313/hematology-causes-differential-secondary-thrombocytosis
Toyama K, Karasawa M, Yamane A, Irisawa H, Yokohama A, Saitoh T et al. JAK2-V617F mutation analysis of granulocytes and platelets from patients with chronic myeloproliferative disorders: advantage of studying platelets. Br J Haematol 2007; 139: 64–69.
Cazzola M, Kralovics R. From Janus kinase 2 to calreticulin: the clinically relevant genomic landscape of myeloproliferative neoplasms. Blood 2014; 123: 3714–3719.
Avraham Z. Cooper. Why does chronic alcohol use cause macrocytosis? https://twitter.com/AvrahamCooperMD/status/1480198409894211589
Jen Wei Ying, Chee Yen Lin and Cheong May Anne. Causes of Macrocytosis https://www.learnhaem.com/courses/anaemia/lessons/approach-to-anaemia/topic/macrocytosis-causes/
Morishita E. [Diagnosis and treatment of microangiopathic hemolytic anemia]. Rinsho Ketsueki. 2015 Jul;56(7):795-806. Japanese. doi: 10.11406/rinketsu.56.795. PMID: 26251142.
Hanif H, Shrestha B, Munankami S, Shrestha M, Poudel B, Reddy R, Jaleel S, Powell D. Severe Malaria with a Rare Tetrad of Blackwater Fever, Acute Renal Failure, Disseminated Intravascular Coagulopathy, and Acute Acalculous Cholecystitis. Case Rep Infect Dis. 2023 May 2;2023:5796881. doi: 10.1155/2023/5796881. PMID: 37179741; PMCID: PMC10169240.
Killick SB, Bown N, Cavenagh J, et al; British Society for Standards in Haematology. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2016;172:187-207.
Bacigalupo A, Hows J, Gluckman E, et al. Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA working party. Br J Haematol. 1988;70:177-182.
What is von Willebrand Disease? https://www.cdc.gov/ncbddd/vwd/facts.html
Von Willebrand Disease. An overview of symptoms and treatments to help you understand VWD. https://www.hemophilia.org/bleeding-disorders-a-z/types/von-willebrand-disease
Kottke-Marchant K. Laboratory diagnosis of hemorrhagic and thrombotic disorders. Hematol Oncol Clin North Am. 8: 1994; 809-853.
Keywords:
mrcp examination requirements, mrcp part 1 examination, mrcp exam eligibility, mrcp exam preparation, mrcp exam questions, mrcp examination dates, mrcp test requirements, how to prepare for mrcp exam, mrcp exam example, mrcp exam explained.









My absolute monocytes count is high range from 8.5 – 15.5 with normal WBC count( 7-9)
This elevation in monocytes absolute count is persisting since more than two years.
I have Graves’ disease
But my mother was diagnosed with CMML converted to CML and she passed away 12 years ago.
For your advice
Hi M Shafei,
Thank you for your comment.
It seems there might be some discrepancy in the information provided.
I recommend emailing a clear copy of your most recent CBC to [email protected] for further review.
Best regards.
Dr M Abdou
Respected,
I have had iron deficiency anemia for years, sometimes less, sometimes more pronounced. (Female, 39 years old, cycles heavier at 25-26 days, one twin pregnancy 9 years ago).
I also drink concor cor (heart failure 1st degree) and eutirox (hashimoto’s) for blood pressure.
The last time my hemoglobin was fine was March 2021 (123), but the constants were still lowered (MCV, MCH, MCHC), as always, Platelets are occasionally elevated. From then on, I was told to take sideral folico regularly for 5 days during the cycle (21 mg of iron, folic acid, vitamin C, D, B6 and B12 together in a sachet), which I respected for a while, but then not. In 2022, the hemoglobin was 110-112. I used to take two sideral folics per case when I had my cycle, and from July of this year until November, I didn’t take any at all. In October of this year, after a month of stomach problems (the doctor said that gastritis, helico and everything else was negative), on a random check the hemoglobin was very low (94). The doctor gave me globifer (18mg), which I took for about a month. After that, the results were almost the same. I switched to sideral folico, and changed my diet a bit – foods rich in iron daily). Also, after a long inactive period, I started a brisk walk every day for 30 minutes in 2 periods (at least an hour of walking in total)
I wanted to check the blood test again after the menstrual cycle, but the results are again more or less the same. I am very worried, because I expected it to be at least a little better, (because in total I take iron for a month and a half, but according to the advice of Dr. NOT some stronger preparation that I took before (tothema, heferum, referral…) than those who have only a daily intake of 18-21 mg, because they say it’s a chronic condition for me)
Could you give me your opinion, what else I could check and change? Is there a suspicion of another disease, or is it just anemia? Otherwise, I get short of breath during exertion, for example, a fast walk, etc. and I have the need to take deep breaths, but the cardiologist told me it was due to heart failure. I have no other complaints. (non-smoker, I don’t consume alcohol, I have extra kg, all other tests are fine – blood fats, bile tests, etc.)
I am asking for your honest advice and opinion. Before taking iron (1.11, – HG 94, HCT 30.2 MCV 67.8 MCHC 311, platelets 476)
the second after taking globifer for 28 days and one abundant cycle (4.12, – HG 94, HCT 31.4 MCV 63.1 MCHC 199, platelets 479)
Today, after 2 weeks of sideral folico and honey with nettle, at the same time a week after a slightly less abundant cycle and a weaker viral infection (nose, throat, cough) (12/19 – HG 94, HCT 31.6 MCV 67.3 MCHC 297, platelets 565).
I am asking for your honest professional opinion and advice. Thanks a lot.
Best Regards!
Hi Ana,
Thank you for reaching out.
The primary cause of iron deficiency anemia in women within your age group often stems from heavy menstrual periods, leading to significant blood loss.
Anemia can manifest with a range of symptoms, such as fatigue, shortness of breath upon exertion, palpitations, and headaches.
I recommend conducting a thorough assessment by checking your serum iron, transferrin saturation, ferritin, reticulocyte count, and Hb electrophoresis.
Hb electrophoresis could help ruling out other causes of anemia like beta thalassemia trait.
Best regards,
Dr. M Abdou