Qualitative Platelet Disorders

Qualitative Platelet Disorders are a critical category of bleeding disorders where the platelets themselves don’t work correctly, resulting in poor clotting function. This happens even when the total platelet count and standard coagulation tests are normal. These disorders cause clinical problems like abnormal bruising or excessive bleeding, and while they are most commonly acquired, some are inherited conditions. Diagnosis often relies on specialized platelet function testing such as light transmission aggregometry (LTA) or the platelet function analyzer (PFA-100), which reveals the underlying defect.
Qualitative platelet disorders are suggested by a prolonged bleeding time (abnormal platelet function screen) or clinical evidence of bleeding in the setting of a normal platelet count and coagulation studies.
The new platelet function test, PFA-100 (Dade-Behring, Deerfield, Ill), has a 96% sensitivity for detecting von Willebrand disease and aspirin-induced platelet defects. It has yet to find a place among routine coagulation laboratory tests.
Drug-Induced Platelet Dysfunction:
Qualitative platelet disorders could be drug-induced. Drug-induced platelet dysfunction is an important cause of an acquired bleeding diathesis. Most commonly, drug-induced platelet dysfunction is considered in a bleeding patient with a normal platelet count, normal screening coagulation tests, and normal renal and liver function. Of course, drugs can exacerbate the bleeding in patients with pre-existing bleeding disorders.
Always obtain a good medication history (prescription, over the counter, herbs and other supplements that patients do not consider as drugs) in every bleeding patient.
The most common drug responsible is aspirin, which irreversibly inhibits cyclooxygenase for 5 to 7 days—hence, the need to hold aspirin for 5 days before elective surgery. Other common drugs include clopidogrel, ticlopidine, and glycoprotein IIb/IIIa inhibitors. Nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit cyclooxygenase reversibly. Ethanol use and aspirin therapy have synergistic effects. Table 1 lists drugs associated with platelet dysfunction. Treatment includes discontinuation of the offending drug and platelet transfusion in the setting of clinically significant bleeding. 
Uremia:
Uremic bleeding syndrome is a recognized consequence of renal failure and can result in clinically significant sequelae. This imparts a predisposition to bleeding that is incompletely understood. As a result of the multifactorial nature of this syndrome, prevention and treatment options can include one or a combination of the following: correction of anemia, dialysis, erythropoietin, cryoprecipitate, desmopressin, and conjugated estrogens. Platelet transfusions do not correct the coagulopathy because the transfused platelets will assume the dysfunction of the uremic platelets.
Liver Disease:
Individuals with liver disease have a variety of hemostatic abnormalities, resulting in “rebalanced” hemostasis. These changes increase risks of bleeding as well as thrombosis. They become especially important in patients with bleeding and thrombotic risks such as gastrointestinal varices and vascular stasis, respectively.
Whether acute or chronic, hepatic disease is associated with platelet dysfunction that is multifactorial in origin. Increased FDP levels from activation of the fibrinolytic pathway compromise platelet function and impair release of platelet factor III from platelets because of cirrhosis or manifestations of hepatic dysfunction.
Acquired von Willebrand Disease:
Acquired von Willebrand disease (AvWD) is an acquired bleeding disorder which may suddenly become manifest in individuals, usually in the absence of a personal or family history of bleedings and frequently in association with monoclonal gammopathies, lymphoproliferative, myeloproliferative and autoimmune disorders. In a minority of the cases AvWD may develop in association with drugs or solid tumours.
Pathogenetic mechanisms involve autoantibodies directed against von Willebrand factor (vWF) resulting in a rapid clearance of vWF from the circulation and/or inactivation of plasma vWF; absorption or adsorption of plasma vWF to malignant cells; drug-induced or cell-mediated proteolysis of plasma vWF; acquired decrease in synthesis of vWF and/or release of vWF from storage sites; or precipitation of plasma vWF.
Treatment options include–whenever possible–treatment of the underlying disorder or symptomatic treatment aimed at replacing the loss of vWF by either infusion of vWF-rich concentrates or administration of desmopressin (DDAVP). In selected cases with anti-vWF antibodies, administration of high-dose intravenous gammaglobulin, plasma exchange or recombinant factor VIIa, depending on availability and urgency.
Paraneoplastic Platelet Dysfunction:
Platelet dysfunction has also been associated with plasma cell dyscrasias and is believed to be related to coating of the platelet membrane by monoclonal proteins. Myelodysplastic and myeloproliferative syndromes may result in platelet dysfunction (e.g., through an acquired glycoprotein IIb/IIIa deficiency). The bleeding time is often prolonged but does not correlate with the bleeding tendency.
Other conditions associated with qualitative platelet disorders include cardiopulmonary bypass or valvular defects, autoimmune disorders (e.g., systemic lupus erythematosus, rheumatoid arthritis, scleroderma), and severe iron or folate deficiency.
Inherited Platelet Disorders
These include the common von Willebrand disease and the less common Glanzmann thrombasthenia and Bernard-Soulier syndrome. The latter two are beyond the scope of this chapter.
Von Willebrand Disease:
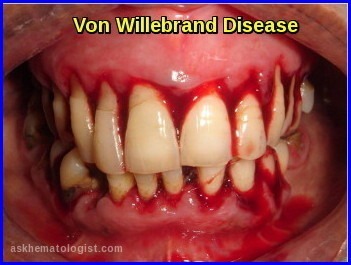
Von Willebrand disease (vWD) is the most common inherited bleeding disorder. It affects about 1% of the population, although only a fraction come to medical attention, often because of the paucity of symptoms in the absence of significant hemostatic challenge but also because of failure to recognize abnormal bleeding. Laboratory testing for mild disease is often difficult to interpret, making its definitive incidence difficult to determine.
People with vWD either have a deficiency of a blood protein called Von Willebrand factor (vWF), or this protein doesn’t work properly. This means that their blood cannot clot properly and they are prone to prolonged or excessive bleeding. Von Willebrand factor is required for platelet adhesion. It binds factor VIII, a key clotting protein, and platelets in blood vessel walls, which help form a platelet plug during the clotting process.
Pathophysiology and Types
vWD is subdivided into three types, based on clinical and laboratory features

Type 1
This accounts for approximately 70% of patients, has an autosomal dominant inheritance, and represents a quantitative deficiency of vWF. Bleeding can be mild to moderately severe.
Type 2
This is further subdivided into the following subtypes:
- Type 2A accounts for about 15% of patients. It is transmitted in an autosomal dominant pattern and involves a deficiency of the high-molecular-weight multimers of vWF. Patients present with moderate to severe bleeding.
- Type 2B accounts for approximately 5% of patients. It is inherited in an autosomal dominant pattern and involves a gain of function mutation that results from increased binding of vWF to platelet glycoprotein 1B and resultant decreased circulating vWF. The hallmark of type 2B vWD is an enhanced aggregation of the patient’s platelets in the presence of ristocetin. Patients with type 2B often have mild thrombocytopenia. Bleeding is moderate to severe.
- Type 2M is rare, autosomal dominant disorder characterized by reduced binding of vWF to platelet glycoprotein 1B.
- Type 2N is a rare, autosomal recessive disorder characterized by decreased binding of vWF to factor VIII, resulting in low factor VIII levels and bleeding patterns similar to those seen in the hemophilias.
Type 3
This is a rare, autosomal recessive subtype, characterized by a marked decrease in vWF. It may result from different genetic defects in compound heterozygotes.
Treatment of Von Willebrand Disease
Treatment of vWD is difficult to monitor because of the lack of laboratory tests that correlate with bleeding. Hence, commonly monitored parameters include clinical bleeding, factor VIII levels, and ristocetin cofactor levels.
Desmopressin
Desmopressin (DDAVP) promotes the release of vWF from endothelial cells. It is effective for patients with type 1 disease, but has a varying effect for patients with type 2A disease. It is relatively contraindicated in patients with type 2B disease. It may also be helpful for patients with type 2M or 2N vWD, but is not helpful for patients with type 3 disease. Desmopressin can be given intra- venously or subcutaneously at 0.2 μg/kg (maximum dose, 20 mg), with a response noted as early as 30 minutes later, lasting 6 to 12 hours. The dose may be repeated in 12 hours and then daily. The intranasal preparation is given at a dose of 150 mg for patients weighing less than 50 kg and 300 mg for those weighing more than 50 kg. A trial infusion is needed to assess the efficacy of treatment and adequacy of prophylactic use. Adverse effects include facial flushing, headaches, hyponatremia with continuous use, and a potential for thrombotic events.
Factor VIII Concentrates
Intermediate purity factor VIII concentrates are used for patients who do not benefit from desmopressin and for those with serious bleeding or before major surgery. Intermediate-purity factor VIII concentrate is used to maintain factor VIII levels between 50% and 100% for 3 to 10 days. A dose of 20 to 30 IU/kg is typically used twice daily. Overzealous treatment results in high factor VIII levels, which is believed to increase the risk of thrombosis.
Aminocaproic Acid and Tranexamic Acid
Aminocaproic acid, 50 mg/kg four times daily, and tranexamic acid, 25 mg/kg three times daily, have been used for mild bleeding episodes and for dental procedures. They carry a risk of thrombotic events, which is especially pronounced in older individuals and with long-term use.
Topical Treatment
Topical treatment for oral or nasal bleeding with Gelfoam or Surgicel soaked with thrombin has been used successfully. Topical therapy plays an important role in prophylaxis and treatment after dental procedures.
Recombinant Factor VIIa
Recombinant factor VIIa has been used successfully in patients with type 3 vWD with alloantibodies. In addition, its use should be considered in patients with life-threatening bleeding in whom other measures have failed. Disadvantages include its high cost as well as increased risks of thrombotic events, which are more pronounced in older adults.
Glanzmann thrombasthenia:
Glanzmann thrombasthenia (GT) is a rare autosomal recessive bleeding syndrome affecting the megakaryocyte lineage and characterized by lack of platelet aggregation. The molecular basis is linked to quantitative and/or qualitative abnormalities of αIIbβ3 integrin. This receptor mediates the binding of adhesive proteins that attach aggregating platelets and ensure thrombus formation at sites of injury in blood vessels.
GT is associated with clinical variability: some patients have only minimal bruising while others have frequent, severe and potentially fatal hemorrhages. The site of bleeding in GT is clearly defined: purpura, epistaxis, gingival hemorrhage, and menorrhagia are nearly constant features; gastrointestinal bleeding and hematuria are less common. In most cases, bleeding symptoms manifest rapidly after birth, even if GT is occasionally only diagnosed in later life.
Diagnosis should be suspected in patients with mucocutaneous bleeding with absent platelet aggregation in response to all physiologic stimuli, and a normal platelet count and morphology. Platelet αIIbβ3 deficiency or nonfunction should always be confirmed, for example by flow cytometry.
In order to avoid platelet alloimmunisation, therapeutic management must include, if possible, local hemostatic procedures and/or desmopressin (DDAVP) administration. Transfusion of HLA-compatible platelet concentrates may be necessary if these measures are ineffective, or to prevent bleeding during surgery. Administration of recombinant factor VIIa is an increasingly used therapeutic alternative. GT can be a severe hemorrhagic disease, however the prognosis is excellent with careful supportive care.
Bernard-Soulier syndrome:
Bernard-Soulier syndrome (BSS), also known as Hemorrhagiparous thrombocytic dystrophy, is a hereditary bleeding disorder affecting the megakaryocyte/platelet lineage and characterized by bleeding tendency, giant blood platelets and low platelet counts.

Peripheral blood smear is diagnostic for Bernard-Soulier syndrome (BSS), demonstrating characteristic giant platelets, which may rival red blood cells in size.
This syndrome is extremely rare as only ~100 cases have been reported in the literature.
Clinical manifestations usually include purpura, epistaxis, menorrhagia, gingival and gastrointestinal bleeding. The syndrome is transmitted as an autosomal recessive trait.
The underlying defect is a deficiency or dysfunction of the glycoprotein GPIb-V-IX complex, a platelet-restricted multisubunit receptor required for normal primary hemostasis. The GPIb-V-IX complex binds von Willebrand factor, allowing platelet adhesion and platelet plug formation at sites of vascular injury. Genes coding for the four subunits of the receptor, GPIBA, GPIBB, GP5 and GP9, map to chromosomes 17p12, 22q11.2, 3q29, and 3q21, respectively. Defects have been identified in GPIBA, GPIBB, and GP9 but not in GP5.
Diagnosis is based on a prolonged skin bleeding time, the presence of a small number of very large platelets (macrothrombocytopenia), defective ristocetin-induced platelet agglutination and low or absent expression of the GPIb-V-IX complex. Prothrombin consumption is markedly reduced.
The prognosis is usually good with adequate supportive care but severe bleeding episodes can occur with menses, trauma and surgical procedures.
Treatment of bleeding or prophylaxis during surgical procedures usually requires platelet transfusion.
References:
Rachid Baz, Tarek Mekhail. Disorders of Platelet Function and Number. The Cleveland Clinic Foundation, Center for Continuing Education. ClevelandClinicMedEd.com.
Mammen E.F., Comp P.C., Gosselin R. (1998). PFA-100 System: A New Method for Assessment of Platelet Dysfunction. Seminars in Thrombosis and Hemostasis, 24: 195–202.
Weigert A.L., Schafer A.I. (1998). Uremic Bleeding: Pathogenesis and Therapy. American Journal of the Medical Sciences, 316: 94–104.
Hedges S.J., Dehoney S.B., Hooper J.S., et al. (2007). Evidence-Based Treatment Recommendations for Uremic Bleeding. Nature Reviews Nephrology, 3: 138–153.
Northup P.G., Caldwell S.H. (2013). Coagulation in Liver Disease: A Guide for the Clinician. Clinical Gastroenterology and Hepatology, 11: 1064–1074.
Kottke-Marchant K. (1994). Laboratory Diagnosis of Hemorrhagic and Thrombotic Disorders. Hematology/Oncology Clinics of North America, 8: 809–853.
George J.N., Caen J-P., Nurden A.T. (1990). Glanzmann’s Thrombasthenia: The Spectrum of Clinical Disease. Blood, 75: 1383–1395.
Lopez J.A., Andrews R.K., Afshar-Kharghan V., Berndt M.C. (1998). Bernard–Soulier Syndrome. Blood, 91: 4397–4418.
Keywords:
Qualitative platelet disorders, platelet disorders, defective platelet function, bleeding disorders platelet, inherited platelet disorders, acquired platelet disorders, platelet function testing, Glanzmann’s thrombasthenia, Bernard-Soulier syndrome, platelet function defects, platelet function abnormalities, bleeding symptoms normal platelet count, platelet function test PFA-100, platelet function aggregometry, treatment qualitative platelet disorders


Related Posts:





Request Online Consultation With Dr M Abdou
Fee: US$100
Secure payment via PayPal (credit and debit cards accepted)
Pay Now