Sickle Cell Disease

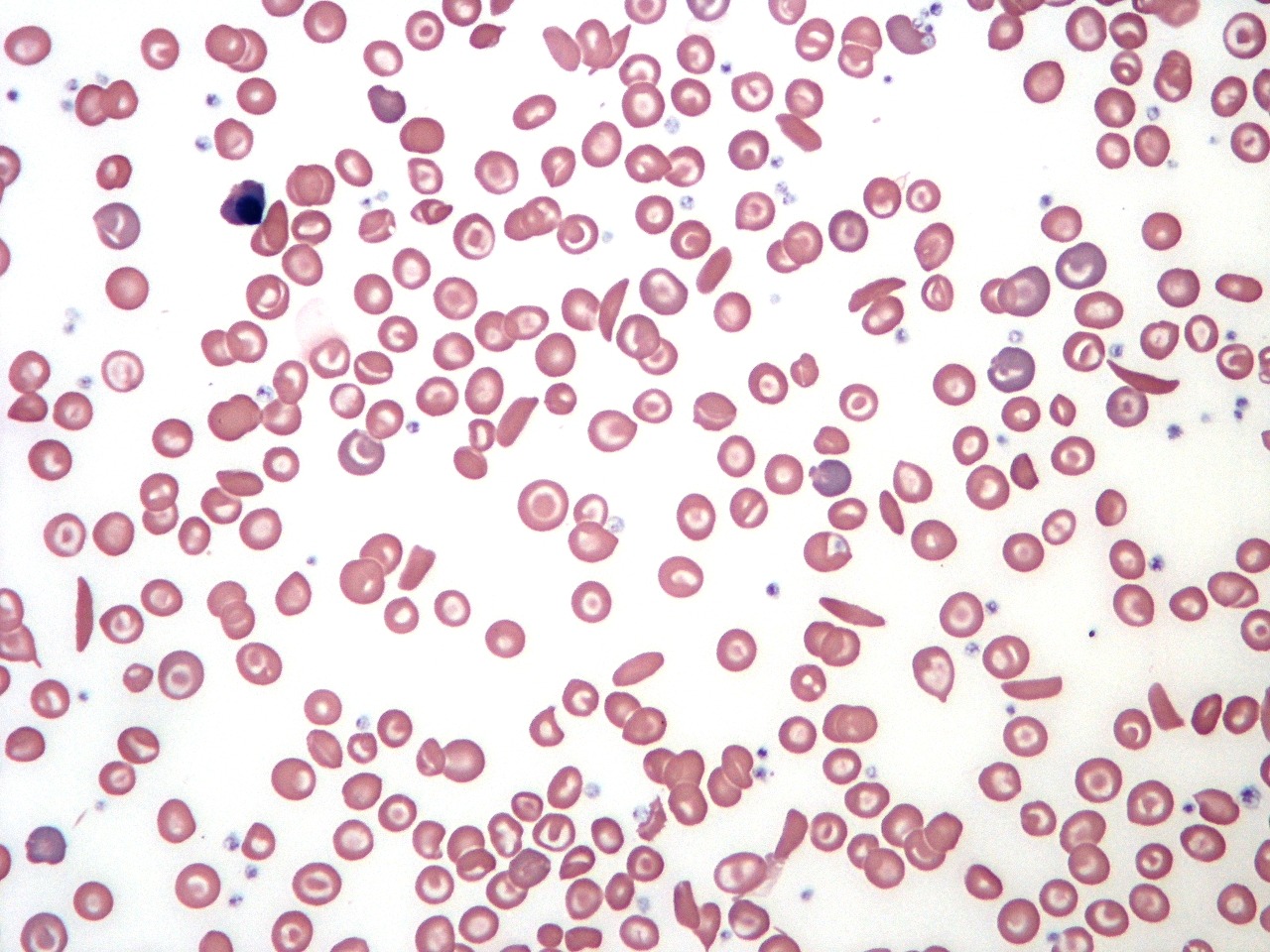
Sickle cell disease blood film demonstrating sickled red cells, target cells, and marked poikilocytosis.
Sickle cell disease (SCD) is a hereditary hemoglobinopathy caused by a point mutation in the β-globin gene, resulting in the production of hemoglobin S (HbS). This abnormal hemoglobin polymerizes under low–oxygen conditions, distorting red blood cells into rigid, crescent-shaped “sickle” forms. Sickle cell disease is most prevalent among individuals of African descent, although it also affects populations in the Middle East, India, and the Mediterranean basin.
The substitution of valine for glutamic acid at the 6th position of the β-globin chain leads to the formation of long, reversible polymers that promote red cell deformation. Sickled erythrocytes become less deformable, more adhesive, and more prone to hemolysis. As they accumulate within the microcirculation, these cells obstruct blood flow, reduce oxygen delivery, and trigger recurrent vaso-occlusive episodes. Endothelial dysfunction, diminished nitric oxide bioavailability, and increased cellular adhesion further exacerbate microvascular occlusion and the chronic complications characteristic of SCD.
Clinical features:
The heterozygous state (Sickle Cell Trait – HbAS) is usually asymptomatic, although problems may arise with anesthesia (hypoxia). People who inherit one sickle cell gene and one normal gene have sickle cell trait (SCT).
People with SCT usually do not have any of the symptoms of sickle cell disease (SCD), but they can pass the trait on to their children. People with sickle cell trait are well, and will usually only know about their trait if they are tested for it. Pregnant women and couples planning children may want to know whether they have sickle cell trait, because if both parents have it, their child might inherit SCD. In England, tests for sickle cell trait and SCD are offered to pregnant women and newborn babies.
There is no anemia and in general, people with sickle cell trait enjoy normal life spans with no medical problems related to sickle cell trait. Rarely, extreme conditions such as severe dehydration and high-intensity physical activity can lead to serious health issues, including sudden death, for individuals with sickle cell trait.
Sickle cell disease (SCD – HbSS) usually presents after 6 months of age, as HbF recedes. There are:
- Vaso-occlusive crises with associated infarct pain affecting particularly the bones, joints, and abdominal organs. Aseptic femoral necrosis, dactylitis, renal damage, priapism and retinopathy all may occur. Dactylitis is severe pain that affects the bones of the hands, the feet, or both. It’s often the first symptom of sickle cell disease in babies caused by blocked blood circulation. Symptoms of dactylitis include extreme pain and tenderness, usually with swelling. An episode of dactylitis may last 1 to 4 weeks. Triggers of the vaso-occlusive crisis include Hypoxemia, Dehydration: Acidosis, and Infections.

Dactylitis in sickle cell disease presenting with acute swelling and tenderness of the hand.

Dactylitis in sickle cell disease with painful swelling of the fingers and hand.

Hand-Foot Syndrome (dactylitis) in sickle cell disease with characteristic swelling of the hands and feet.
- Anemia may be due to hemolysis, ‘aplastic crises‘ precipitated by parvovirus B19 infection, folate deficiency, and ‘sequestration crises‘ in the liver or spleen.
- Splenomegaly is common in childhood but disappears as the spleen is infarcted. Patients with SCD who have damaged spleens are at risk for serious bacterial infections that can be life-threatening. Some of these bacteria include Pneumococcus, Hemophilus influenza type B, Meningococcus, Salmonella, Staphylococcus, Chlamydia, and Mycoplasma pneumoniae.
- Leg ulceration.

Chronic leg ulcer associated with sickle cell disease, showing tissue loss and hyperpigmented surrounding skin.
- Acute chest syndrome: Sickled RBCs in the blood vessels of the lungs can cause pulmonary infarcts. This condition is very serious and should be treated immediately at a hospital. Acute chest syndrome often starts a few days after a painful crisis begins. A lung infection may accompany acute chest syndrome. CT pulmonary angiography (CTPA) is indicated in suspected cases of pulmonary embolism (PE).
- Pulmonary hypertension: increasingly recognized as a serious complication of SCD.
- Avascular necrosis of the femoral or humeral head due to vascular occlusion.
- CNS involvement, e.g. stroke.
- Ophthalmologic involvement: ptosis, retinal vascular changes, proliferative retinitis.
- Cardiac involvement: dilation of both ventricles and the left atrium.
- GI involvement: cholelithiasis is common in children; liver may become involved.
- GU involvement: kidneys lose concentrating capacity.
Investigations:
The blood appears normal in sickle cell trait, although the sickle screening test is positive.
Sickle cell disease is suggested by the typical clinical picture of chronic hemolytic anemia with reticulocytes and sickle cells on the peripheral blood film, and vaso-occlusive crisis. Electrophoresis confirms the diagnosis with the presence of homozygous HbSS (no HbA) and can also document other hemoglobinopathies (eg, HbSC, HbS-beta+ thalassemia).
Laboratory tests used in patients with SCD include the following:
- FBC with differential and reticulocyte count.
- Peripheral blood smear.

Comparison of a sickle cell disease blood film showing sickled erythrocytes, target cells, and nucleated red blood cells (NRBCs) versus a normal film with regular red cell morphology.
- Hb electrophoresis.
- Hemoglobin solubility testing.
- Serum electrolytes.
- Renal function (creatinine, urea, urinalysis).
- LFTs (ALT, indirect and direct bilirubin).
- Pulmonary function tests (transcutaneous O 2 saturation).
- ABGs.
- Blood cultures.
- CSF examination: Consider LP in febrile children who appear toxic and in those with neurologic findings (eg, neck stiffness, + Brudzinski/Kernig signs, focal deficits); consider CT brain scanning before performing LP.
- Secretory phospholipase A2.
N.B. Mandatory screening for HbS at birth in the United States; prenatal testing can be obtained via chorionic villus sampling.
Management:
- Prevention of Crises: patients with SCD should avoid dehydration, hypoxia, acidosis or cold. Infections should be treated promptly.

Common triggers of sickle cell crises include infections, low oxygen tension, dehydration, acidosis, extreme exertion, stress, alcohol, pregnancy, and exposure to cold.

Key treatment goals in sickle cell disease: managing vaso-occlusive crises, chronic pain and hemolysis, preventing infections and organ damage, and addressing risks of stroke and pulmonary hypertension.
- Crises are treated by rest, rehydration, management of underlying infection and complications, and administration of analgesics. Oxygen supplementation helps only if the patient has hypoxia. Rapid initiation of opioids for the treatment of severe pain associated with a vaso-occlusive crisis is highly recommended. Patient-controlled analgesia (PCA) is a method of safely administering strong opioids which is controlled by the patient (or a nurse for nurse-controlled analgesia).

Oral analgesics commonly used in adult vaso-occlusive crisis, detailing recommended dosing for opioids and non-opioid agents.
- Blood transfusion is indicated in severely affected individuals. Preoperative transfusion therapy should be used to increase hemoglobin levels to 10 g/dL. With continued transfusion, iron overload inevitably develops and can result in heart and liver failure and multiple other complications. Deferasirox (Exjade) has a capacity similar to desferrioxamine (Desferal) in chelating iron, but it is administered orally. Renal toxicity might be a limiting factor in its use, but it is generally safe.
- Folic acid supplements.
- Daily oral prophylactic penicillin (or erythromycin if allergic to penicillin) up to age 5. Many hematologists prefer to continue this antibiotic throughout life in patients with SCD (HbSS).
- There is a strong evidence supporting the efficacy of Hydroxyurea in adults with SCD. It can decrease severe painful episodes, hospitalizations, the frequency of blood transfusions, and the acute chest syndrome. Although the evidence for the efficacy of hydroxyurea treatment for children is not as strong, the emerging data are encouraging. Hydroxyurea increases total and fetal hemoglobin (HbF) in children with SCD. The increase in HbF retards sickling of RBCs. Hydroxyurea also reduces levels of circulating leucocytes, which decreases the adherence of neutrophils to the vascular endothelium. In turn, these effects reduce the incidence of pain episodes and acute chest syndrome episodes.
- Allogeneic Bone Marrow Transplantation (BMT) may be justified in severely affected children, particularly those who have had cerebrovascular incidents.
- Immunization: People with SCD should receive all recommended childhood vaccines. They should also receive additional vaccines to prevent other infections (pneumococcus, meningococcus, influenza).

Recommended immunization schedule for sickle cell disease, including Haemophilus influenzae, meningococcal, pneumococcal, hepatitis B, and tetanus booster vaccinations.
Other Sickling Disorders
In addition to classic sickle cell disease, several related hemoglobinopathies can produce sickling phenomena. Sickle β-thalassemia occurs when the sickle cell mutation is inherited alongside a β-thalassemia allele, leading to variable disease severity depending on whether the β-thalassemia defect is mild (β⁺) or severe (β⁰).
Hemoglobin C disease and the compound heterozygous state HbSC may also result in intermittent sickling. Individuals with HbSC often experience mild to moderate hemolytic anemia, splenomegaly, and are particularly prone to ocular complications, including proliferative retinopathy.
Summary:
Sickle cell disease is an inherited hemoglobinopathy in which abnormal hemoglobin S causes red blood cells to become rigid, fragile, and sickle-shaped. These distorted cells obstruct small blood vessels, leading to recurrent vaso-occlusive crises, chronic hemolytic anemia, impaired oxygen delivery, and progressive organ damage. Individuals inherit two abnormal hemoglobin genes, and the resulting disease manifests with acute pain episodes, fatigue, increased susceptibility to infections, leg ulcers, stroke risk, and other systemic complications.
Ongoing medical management is essential, including pain control, infection prophylaxis, hydroxyurea therapy, blood transfusion programs, and in selected cases, stem cell transplantation. Early diagnosis through newborn screening, patient education, and genetic counselling significantly improves long-term outcomes. Continued research into disease-modifying therapies and potential curative approaches, including gene-based treatments, offers growing hope for individuals living with sickle cell disease.
Questions and Answers:
What is sickle cell disease and how does it affect red blood cells?
Sickle cell disease is an inherited hemoglobinopathy in which abnormal hemoglobin S causes red blood cells to become rigid, sickle-shaped, and prone to hemolysis, leading to vaso-occlusion, impaired oxygen delivery, and organ damage.
What causes sickle cell crises and why do they happen?
Sickle cell crises occur when sickled red cells obstruct the microvasculature during triggers such as infection, dehydration, low oxygen tension, acidosis, cold exposure, stress, or extreme physical exertion.
What are the most common symptoms of sickle cell disease?
Common symptoms include recurrent vaso-occlusive pain episodes, chronic anemia, fatigue, jaundice, susceptibility to infections, dactylitis in infants, acute chest syndrome, stroke, and multi-organ complications.
How is sickle cell disease diagnosed?
Diagnosis is confirmed using hemoglobin electrophoresis or high-performance liquid chromatography (HPLC), supported by blood film findings such as sickled cells, target cells, NRBCs, and evidence of chronic hemolysis.
What is dactylitis in sickle cell disease?
Dactylitis is the early manifestation of sickle cell disease, presenting as painful swelling of the hands and feet in infants due to microvascular occlusion and bone marrow infarction.
What complications can occur in sickle cell disease?
Complications include acute chest syndrome, splenic sequestration, stroke, leg ulcers, avascular necrosis, retinopathy, pulmonary hypertension, kidney disease, and recurrent infections due to functional asplenia.
How is sickle cell pain managed?
Pain management involves hydration, NSAIDs, acetaminophen, and opioid analgesics for severe crises, alongside disease-modifying therapy such as hydroxyurea to reduce frequency and severity of vaso-occlusive events.
What is acute chest syndrome in sickle cell disease?
Acute chest syndrome is a life-threatening complication characterized by chest pain, fever, respiratory symptoms, and new pulmonary infiltrates caused by infection, fat embolism, or in situ vaso-occlusion.
How does hydroxyurea help patients with sickle cell disease?
Hydroxyurea increases fetal hemoglobin (HbF) levels, reduces sickling, decreases frequency of vaso-occlusive crises, lowers the risk of acute chest syndrome, and improves overall survival.
What is the role of blood transfusions in sickle cell disease?
Blood transfusions are used to treat severe anemia, reduce stroke risk, manage acute chest syndrome, and prevent complications by lowering circulating HbS levels.
Can sickle cell disease be cured?
Curative options include allogeneic hematopoietic stem cell transplantation and emerging gene-based therapies, though availability and eligibility depend on age, disease severity, and donor match.
What lifestyle measures help reduce sickle cell complications?
Adequate hydration, avoiding extreme temperatures, preventing infections, minimizing stress, prompt pain management, and adherence to medical therapy all reduce the frequency and severity of crises.
What are the ocular complications of HbSC disease?
HbSC disease is particularly associated with proliferative sickle retinopathy, which can cause visual loss due to neovascularization and retinal detachment.
How do sickle cell ulcers develop?
Leg ulcers occur due to microvascular obstruction, chronic hemolysis, impaired oxygen delivery, and local tissue ischemia, often becoming chronic and slow to heal.
What is sickle β-thalassemia?
Sickle β-thalassemia is a compound hemoglobinopathy where HbS is inherited with a β-thalassemia allele, producing a variable phenotype ranging from mild disease (β⁺) to severe disease (β⁰).
References:
Wang WC. Sickle cell anemia and other sickling syndromes. In: Greer JP, et al., editors. Wintrobe’s Clinical Hematology. 12th ed. Philadelphia: Lippincott Williams & Wilkins; 2009. p. 1038–1082.
Yawn BP, et al. Management of sickle cell disease: Summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(10):1033–1048.
Brawley OW, et al. National Institutes of Health consensus development conference statement: Hydroxyurea treatment for sickle cell disease. Ann Intern Med. 2008;148(12):932–938.
Serjeant GR. Sickle Cell Disease. 2nd revised ed. Oxford: Oxford University Press; 1992.
Ohene-Frempong K, Nkrumah FK. Sickle cell disease in Africa. In: Embury SH, Hebbel RP, Mohandas N, et al., editors. Sickle Cell Disease: Basic Principles and Clinical Practice. New York: Raven Press; 1994.
Olujohungbe A, Howard J. The clinical care of adult patients with sickle cell disease. Br J Hosp Med (Lond). 2008;69(11):616–619.
Onimoe G, Rotz S. Sickle cell disease: A primary care update. Cleveland Clinic Journal of Medicine. 2020;87(1):19–27.
Linguraru MG, Orandi BJ, Van Uitert RL, Mukherjee N, Summers RM, Gladwin MT, et al. CT and image processing non-invasive indicators of sickle cell secondary pulmonary hypertension. In: Conf Proc IEEE Eng Med Biol Soc. 2008;2008:859–862.
Keywords:
sickle cell, sickle cell disease, sickle cell anemia, sickle cell anemia symptoms, sickle cell disease symptoms, sickle cell trait, sickle cell trait symptoms, sickle cell crisis, sickle cell crisis symptoms, sickle cell crisis triggers, vaso-occlusive crisis, sickle cell dactylitis, sickle cell leg ulcers, sickle cell anemia treatment, hydroxyurea sickle cell, sickle cell cure, gene therapy sickle cell, sickle cell anemia genetics, hemoglobin S, hemoglobinopathy, sickle cell anemia life expectancy, sickle cell complications, acute chest syndrome, sickle cell in children, newborn screening sickle cell, sickle cell anemia and malaria, sickle cell trait vs disease, sickle cell pain management, sickle cell crisis management, sickle cell crisis labs, sickle cell infection risk, sickle cell vaccinations, sickle cell pulmonary hypertension, sickle cell retinopathy, sickle beta thalassemia, HbSC disease, sickle cell anemia in adults, chronic hemolytic anemia, sickled red blood cells, target cells NRBCs sickle cell






Thank you for this amazing piece. I learnt a lot from this. Please keep the good work going.
Thank you sir.
Thank you so much sir for detailed information regarding sickle cell disease.
excellent article
many thanks
taj
Hi Taj,
I’m glad to hear that you found the article helpful.
Best wishes,
Dr. M. Abdou