Castleman Disease

Castleman disease histopathology demonstrating the characteristic hyaline vascular variant with concentric “onion-skin” lymphoid follicles and a central hyalinized vessel
What is Castleman Disease?
Castleman disease is a rare lymphoproliferative disorder that affects the lymph nodes and related tissues. It was first described in the 1950s by Benjamin Castleman. The condition is also known as Castleman’s disease, giant lymph node hyperplasia, or angiofollicular lymph node hyperplasia (AFH).
Castleman disease is not a cancer, but it is classified as a non-clonal lymphoproliferative disorder, meaning there is abnormal overgrowth of lymphoid cells. Clinically and biologically, this process shares several features with lymphoma, which can make diagnosis and management challenging.
One subtype, Multicentric Castleman Disease (MCD), behaves in an aggressive manner similar to lymphoma and is associated with systemic inflammation, constitutional symptoms, and immune dysregulation. Patients with MCD have an increased risk of developing lymphoma and are often treated with therapies such as immunotherapy, chemotherapy, or radiation, which is why Castleman disease is discussed within cancer-related resources, including those of the American Cancer Society.
Castleman disease is classified clinically based on the distribution of lymph node involvement. Unicentric Castleman Disease (UCD) affects a single lymph node or a single nodal region, whereas Multicentric Castleman Disease (MCD) involves multiple lymph node regions and systemic manifestations.
MCD is further divided into subtypes. Some cases are caused by Human herpesvirus 8 (HHV-8), also known as Kaposi sarcoma–associated herpesvirus. These cases are termed HHV-8-associated MCD. Other patients are HHV-8 negative, with no identifiable cause; this form is known as idiopathic Multicentric Castleman Disease (iMCD).
Histopathologically, Castleman disease may be described as hyaline-vascular, plasmacytic, mixed, or plasmablastic, based on microscopic lymph node features. However, the clinical utility of this histological sub-classification is limited compared with clinical and virological classification.
Castleman Disease Causes:
It’s not clear what causes Castleman disease. However, infection by a virus called human herpesvirus 8 (HHV-8) is associated with multicentric Castleman disease.
The HHV-8 virus has also been linked to the development of Kaposi’s sarcoma, a cancerous tumor that can be a complication of HIV/AIDS. Studies have found that HHV-8 is present in nearly all HIV-positive people who have Castleman disease, and in about half of HIV-negative people with Castleman disease.
If a patient has multiple regions of enlarged lymph nodes with CD features identified by a pathologist under the microscope AND is HIV-negative and his/her lymph node and blood sample are HHV-8-negative, he/she may have HHV-8-negative or idiopathic MCD (iMCD).
Castleman disease can affect people of any age. But the average age of people diagnosed with unicentric Castleman disease is 35. Most people with the multicentric form are in their 50s and 60s. The multicentric form is also slightly more common in men than in women.
The risk of developing multicentric Castleman disease is higher in people who are infected with HHV-8 virus.
Castleman Disease Signs & Symptoms:
Interleukin 6 (IL6) plays an important role in the pathophysiology of the disease, symptomatology, and as a potential therapeutic target. CD occurs at a relatively younger age with the highest incidence in mid-thirties to forties.
Many people with unicentric Castleman disease don’t notice any signs or symptoms. The enlarged lymph node may be detected during a physical exam or an imaging test for some unrelated problem.

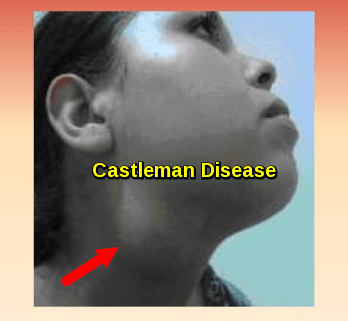
Cervical lymphadenopathy in Castleman disease, showing visible enlargement of neck lymph nodes
Some people with unicentric Castleman disease might experience signs and symptoms more common to multicentric Castleman disease, which may include:
- Fever
- Unintended weight loss
- Fatigue
- Night Sweats
- Nausea
- Enlarged liver or spleen
The enlarged lymph nodes associated with multicentric Castleman disease are most commonly located in the neck, collarbone, underarm and groin areas.
IMCD involves multicentric lymphadenopathy with characteristic “CD-like” lymph node histopathology and a number of signs and symptoms as defined by the 2017 International Consensus Diagnostic Criteria of iMCD, which may progress or remit/relapse over time:
- Elevated C-reactive protein (CRP) level and/or erythrocyte sedimentation rate (ESR)
- Anemia
- Thrombocytopenia or thrombocytosis
- Hypoalbuminemia
- Renal dysfunction and/or proteinuria
- Polyclonal hypergammaglobulinemia
- Flulike symptoms (night sweats, fever, weight loss, fatigue) which are related to high serum IL6 level.
- Large liver and/or spleen
- Fluid accumulation (edema, anasarca, ascites, pleural effusion)
- Eruptive cherry hemangiomatosis or violaceous papules
- Lymphocytic interstitial pneumonitis

Infographic illustrating the major signs and symptoms of Castleman disease, including lymph node enlargement, systemic inflammatory features, and organ involvement
Diagnosis:
Pathologic evaluation of lymph node biopsy is essential for diagnosis. There are two basic types: hyaline-vascular type, and plasmacytic variant, with occasional cases having a mixed picture. In the hyaline-vascular type of the CD, lymph node biopsy shows angiofollicular hyperplasia with depleted germinal center cells and concentric expansion of mantle zones with small lymphocytes (onion skin appearance). The pathology shows hyalinized blood vessels as well as the presence of follicular dendritic cells, but lack of plasma cells. The increased blood vessels are considered to be related to the production of VEGF, which is produced in the presence of and by elevated IL6. Follicular cells seen in hyaline vascular CD express CD20 and CD5 but are not clonal.

Pathologic features of Castleman disease demonstrating the classical hyaline-vascular type with lymphoid hyperplasia, broad mantle zones, and prominent interfollicular vascular proliferation

Hyaline vascular Castleman disease showing hyalinized vessels within an atrophic follicle center on H&E staining
The plasmacytic variant of CD shows an interfollicular increase in polyclonal plasma cells. The lymphoid architecture is relatively preserved but there are hyperplastic follicles. The mixed type of CD shows features representing both hyaline-vascular type and plasmacytic type.

Plasma cell variant of Castleman disease showing reactive lymphoid follicle with hyperplastic germinal center and prominent interfollicular plasma cell infiltrate without significant vascular proliferation
Ninety percent of the unicentric CD is hyaline vascular; while 80 to 90% of the multicentric CD is either plasmacytic or mixed type.
A very small number of patients, usually multicentric plasmacytic variant, may evolve into lymphoma.
The diagnosis of Castleman’s disease should be expected in patients with a localized lymph node enlargement or a generalized lymphadenopathy with typical IL6 related symptoms including fever, night sweats, fatigue, and weight loss.
Additional symptoms as described above may include those due to localized mass or due to the production of cytokines (VEGF). The physical examination will mainly demonstrate the presence of either localized or generalized lymphadenopathy. Also, patients may present with dermatological or neurological manifestations of the disease, or with signs and symptoms associated with autoimmune disorders and/or anemia, if present.
In general, the signs and symptoms associated with CD are non-specific and are not sufficient to establish the diagnosis. There is no single pathognomonic presenting feature, and biopsy of the lymph node and histological confirmation remains the only absolute test for diagnosis. In a number of situations, the investigations are performed to exclude other conditions, especially malignant diseases.
Computed tomography (CT) scan of neck, chest, abdomen, and pelvis is essential for both diagnosing CD and to stage it between unicentric versus multicentric disease. The CT scan will also help identify the presence of hepatosplenomegaly.

Contrast-enhanced CT scan showing an enhancing abdominal mass consistent with Castleman disease adjacent to the head of the pancreas
Positron emission tomography-computed tomography (PET–CT) is also helpful as lymph nodes from CD are positive with low SUV (Standard Uptake Value). It helps differentiate between lymphoma and other malignancies, where the lymph nodes have a much higher SUV.
Differential Diagnosis:
The differential diagnosis of CD is broad and includes disorders that are overlapping with lymphadenopathy, as well as pathological findings.
All the possible causes of localized or generalized lymphadenopathy should be considered as a possible differential diagnosis of CD. The first and the foremost conditions to be excluded are the malignant disorders, specifically non-Hodgkin’s or Hodgkin’s lymphoma, as these diseases require a different and specific therapeutic intervention. None of the laboratory studies are able to differentiate malignant conditions from CD, and lymph node biopsy with appropriate histological, immunohistochemical, and molecular analysis is essential for diagnosis. Other conditions in the differential diagnosis include autoimmune disorders, such as rheumatoid arthritis and connective tissue diseases. Their diagnosis would also depend on both clinical differentiation, as well as various serological testing.
The pathological evaluation and differential diagnosis include most importantly identification and differentiation of reactive lymphadenopathy observed in a number of diseases. This again includes autoimmune diseases such as connective tissue disorders and collagen vascular diseases, HIV and other immunodeficiency related lymphadenopathy, as well as infection etiologies.
Treatment:
For localized (unicentric) disease, surgical removal of the affected lymph node(s) usually results in a cure. However, recurrences of UCD have been reported. In some cases, ionizing radiation (radiotherapy) has proven effective.
For iMCD, chemotherapy had been the cornerstone of treatment but has been largely supplanted by newer, more directed therapies. These include drugs which target and neutralize IL-6 (siltuximab or Sylvant) or the receptor for IL-6 (tocilizumab or Actemra). In 2014, Sylvant (siltuximab) was approved to treat patients with iMCD. This is the first and only FDA-approved drug to treat patients with iMCD. Approximately half of the iMCD patients do not improve with IL-6 neutralization. These patients are often treated with chemotherapy or newer treatment options such as rituximab, sirolimus, or anakinra.
Additional symptomatic and supportive therapy may include corticosteroids or autologous bone marrow transplantation (used most frequently for severe disease or iMCD associated with POEMS syndrome).
For HHV-8-associated MCD, rituximab to eliminate the B lymphocyte is often used. It is highly effective for HHV-8-associated MCD, but occasionally antivirals and/or cytotoxic chemotherapies are needed.
Tocilizumab (Actemra), manufactured by Roche Pharmaceuticals in the United States, showed substantial efficacy in the treatment of iMCD in a small study from Japan. The drug was approved by the FDA for the treatment of rheumatoid arthritis and can be prescribed “off-label” for the treatment of Castleman. Treatment requires monthly intravenous injections.
Prognosis:
The prognosis of CD varies depending on the type and severity of the disease. UCD has an excellent prognosis with surgical excision alone. MCD has a more variable prognosis and can be fatal if left untreated or if there is organ dysfunction.
Summary:
Castleman disease is a rare non-clonal lymphoproliferative disorder that can affect individuals of any age, sex, or ethnicity. It is broadly classified into unicentric Castleman disease (UCD), which involves a single lymph node region, and multicentric Castleman disease (MCD), a systemic inflammatory condition characterized by generalized lymphadenopathy and multi-organ involvement.
Diagnosis relies on a combination of clinical features, laboratory abnormalities, cross-sectional imaging, and excisional lymph node biopsy with characteristic histopathological findings. Management varies according to disease subtype and severity; surgical resection is often curative in UCD, whereas MCD typically requires systemic therapy, including anti-IL-6–targeted agents, immunotherapy, and, in selected cases, chemotherapy.
The prognosis of Castleman disease is heterogeneous and depends on the clinical subtype, underlying etiology (such as HHV-8 status), response to therapy, and presence of complications, with outcomes significantly improved in recent years due to advances in targeted treatment strategies.
Questions and Answers:
What is Castleman disease?
Castleman disease is a rare non-clonal lymphoproliferative disorder characterized by abnormal enlargement of lymph nodes and immune dysregulation. Although it is not a cancer, it can clinically resemble lymphoma and may be associated with an increased risk of lymphoid malignancies.
Is Castleman disease a cancer or lymphoma?
Castleman disease is not classified as cancer or lymphoma; however, multicentric Castleman disease behaves aggressively and shares several clinical and biological features with lymphoma, including systemic inflammation and response to immunochemotherapy.
What are the main types of Castleman disease?
Castleman disease is divided into unicentric Castleman disease, which involves a single lymph node region, and multicentric Castleman disease, which affects multiple lymph node regions and causes systemic inflammatory manifestations.
What is unicentric Castleman disease (UCD)?
Unicentric Castleman disease is a localized form affecting one lymph node or nodal region. Patients are often asymptomatic or present with a painless mass, and complete surgical excision is usually curative.
What is multicentric Castleman disease (MCD)?
Multicentric Castleman disease is a systemic inflammatory disorder characterized by generalized lymphadenopathy, constitutional symptoms, laboratory abnormalities, and multi-organ involvement, requiring systemic medical therapy.
What is the difference between HHV-8-associated and idiopathic multicentric Castleman disease?
HHV-8-associated multicentric Castleman disease is driven by human herpesvirus-8 infection and is more common in immunocompromised patients, whereas idiopathic multicentric Castleman disease is HHV-8 negative and primarily mediated by cytokine dysregulation, particularly excess interleukin-6.
What are the common symptoms of Castleman disease?
Symptoms may include painless lymph node enlargement, fever, night sweats, weight loss, fatigue, anemia, edema, hepatosplenomegaly, and organ dysfunction, especially in multicentric disease.
How is Castleman disease diagnosed?
Diagnosis is based on a combination of clinical features, laboratory abnormalities, cross-sectional imaging such as CT scanning, and confirmatory excisional lymph node biopsy demonstrating characteristic histopathological findings.
What are the histological types of Castleman disease?
The main histological patterns include the hyaline-vascular type, plasma cell variant, mixed type, and plasmablastic variant, although clinical classification is more important than histology alone for prognosis and management.
What are the key histopathologic features of hyaline-vascular Castleman disease?
Hyaline-vascular Castleman disease shows regressed or atrophic germinal centers, concentric mantle zone lymphocytes forming an onion-skin pattern, and hyalinized penetrating blood vessels producing the classic lollipop appearance.
What characterizes the plasma cell variant of Castleman disease?
The plasma cell variant is characterized by hyperplastic germinal centers and dense interfollicular plasma cell infiltration and is often associated with systemic symptoms and inflammatory laboratory abnormalities.
How does Castleman disease appear on CT imaging?
On contrast-enhanced CT, Castleman disease typically appears as a well-defined hypervascular lymph node mass, particularly in unicentric disease, and may mimic malignant or pancreatic masses.
What treatments are used for Castleman disease?
Treatment depends on disease subtype; unicentric Castleman disease is managed surgically, whereas multicentric Castleman disease usually requires systemic therapy including immunotherapy, targeted agents, corticosteroids, and chemotherapy in selected cases.
What targeted therapies are used in multicentric Castleman disease?
Targeted therapies include interleukin-6 pathway inhibitors such as siltuximab and tocilizumab, which have significantly improved outcomes in idiopathic multicentric Castleman disease.
What is the prognosis of Castleman disease?
Prognosis varies by subtype; unicentric Castleman disease has an excellent outcome after surgical resection, while multicentric Castleman disease has a variable prognosis depending on HHV-8 status, response to therapy, and disease severity.
Can Castleman disease recur or progress to lymphoma?
Recurrence can occur, particularly in multicentric disease, and affected patients have an increased risk of developing lymphoma, making long-term follow-up essential.
References:
Munshi N. Castleman’s Disease. Cancer Therapy Advisor.
https://www.cancertherapyadvisor.com/home/decision-support-in-medicine/hematology/castlemans-disease-2/
National Organization for Rare Disorders (NORD). Castleman Disease.
https://rarediseases.org/rare-diseases/castlemans-disease/
American Cancer Society. What Is Castleman Disease?
https://www.cancer.org/cancer/castleman-disease/about/what-is-castleman-disease.html
Fajgenbaum DC. Castleman Disease. National Organization for Rare Disorders (NORD).
https://rarediseases.org/rare-diseases/castlemans-disease/
Mayo Clinic. Castleman Disease – Symptoms and Causes.
https://www.mayoclinic.org/diseases-conditions/castleman-disease/symptoms-causes/syc-20370735
Castleman Disease Collaborative Network (CDCN). About Castleman Disease.
https://www.cdcn.org/about-castleman-disease
Radhakrishnan N. Castleman Disease: Practice Essentials, Pathophysiology, Etiology. Medscape.
https://emedicine.medscape.com/article/2219018-overview
Chang KC, et al. Monoclonality and cytogenetic abnormalities in hyaline vascular Castleman disease. ResearchGate.
https://www.researchgate.net/figure/Pathologic-features-of-Castleman-disease-The-classical-type-of-hyaline-vascular_fig2_258347640
Balakrishna J. Lymph Nodes – Not Lymphoma: Castleman Disease. Pathology Outlines.
http://www.pathologyoutlines.com/topic/lymphnodescastleman.html
Saeed I, Al-Amri AM. Castleman Disease. Korean Journal of Hematology. 2012;47(3):163–177.
doi:10.5045/kjh.2012.47.3.163
DoveMed. Castleman Disease (CD).
https://www.dovemed.com/diseases-conditions/castleman-disease-cd/
Fajgenbaum DC, Uldrick TS, Bagg A, et al. International evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. 2017;129(12):1646–1657.
doi:10.1182/blood-2016-10-746933
van Rhee F, Stone K, Szmania S, Barlogie B, Singh Z. Castleman Disease in the 21st Century: Diagnosis, Assessment, and Therapy. Clinical Advances in Hematology & Oncology. 2018;16(7):482–498.
El-Osta HE, Kurzrock R. Castleman’s Disease: From Basic Mechanisms to Molecular Therapeutics. The Oncologist. 2011;16(4):497–511.
doi:10.1634/theoncologist.2010-0404
Dispenzieri A, Armitage JO, Loe MJ, et al. The clinical spectrum of Castleman’s disease. American Journal of Hematology. 2012;87(11):997–1002.
doi:10.1002/ajh.23283
StatPearls Publishing. Castleman Disease. StatPearls [Internet].
https://www.ncbi.nlm.nih.gov/books/NBK544366/
National Cancer Institute (NCI). Castleman Disease Overview.
https://www.cancer.gov/pediatric-adult-rare-tumor/rare-tumors/rare-hematologic-tumors/castleman-disease
New England Journal of Medicine. Advances in the Understanding and Treatment of Castleman Disease. N Engl J Med. 2020;383(20):1954–1962.
doi:10.1056/NEJMra2025290
Keywords:
Castleman disease, unicentric Castleman disease, multicentric Castleman disease, idiopathic multicentric Castleman disease, HHV-8 associated Castleman disease, lymphoproliferative disorder, Castleman disease symptoms, Castleman disease diagnosis, Castleman disease pathology, Castleman disease histology, hyaline vascular Castleman disease, plasma cell variant Castleman disease, Castleman disease CT scan, Castleman disease imaging, lymph node biopsy Castleman disease, IL-6 mediated inflammation, IL-6 inhibitors Castleman disease, siltuximab Castleman disease, tocilizumab Castleman disease, Castleman disease treatment guidelines, Castleman disease prognosis, Castleman disease vs lymphoma, Ask Hematologist Castleman disease, hematology educational articles

Related Posts:





Request Online Consultation With Dr M Abdou
Fee: US$100
Secure payment via PayPal (credit and debit cards accepted)
Pay Now
report says exactly mentioned below while investigating an incidental finding of gamma globulinemia.High resolution protein electrophoresis shows densely staining monoclonal gammopathy (M spike) in gamma globulin region.Immunofixation shows IgA and lambda.Polyclonal gamma globulin to mainly consists of IgG,kappa,lambda and fair amount of IgA.
Now patient details is Age 78, Hypertriglyceridaemia with Diffuse fatty liver and border line diabetic controlled by diet and discipline.Patients recent complain vertigo only.He had high ESR above 100 and slight anaemia Hb 10.9 with creatinine 1.4.So i went for skeletal survey with normal findings but protein electrophoresis found abnormal gammopathy.So sent for immunofixation that gave the above mentioned report.
So can i get some guidance regarding this patients management?My D/D is MGUS or MGRS.
Hello,
Thank you for your message.
How much the paraprotein quantitation and serum calcium level?
I would recommend bone marrow examination to out rule myeloma.
Best wishes,