Chronic Granulomatous Disease

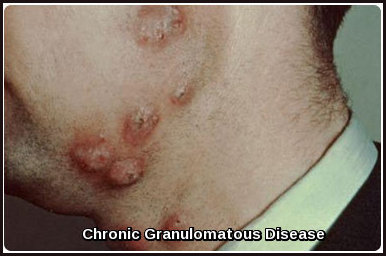
Chronic Granulomatous Disease: cutaneous granulomatous lesions commonly seen in CGD due to impaired phagocyte oxidative burst.
Chronic Granulomatous Disease (CGD) is a rare inherited primary immunodeficiency in which phagocytes fail to generate reactive oxygen species (ROS) due to defects in the NADPH oxidase enzyme complex. As a result, affected individuals are highly susceptible to recurrent, severe bacterial and fungal infections and may develop granulomatous inflammation in various organs. The study of CGD has been pivotal in advancing our understanding of the phagocyte oxidase system and host–pathogen interactions. Most cases are inherited in an X-linked pattern, although autosomal recessive forms also occur, reflecting mutations in different components of the NADPH oxidase pathway.
Affected individuals suffer from recurrent infections, particularly with staphylococci.
CGD is a primary immunodeficiency that affects phagocytes of the immune system and leads to recurrent or persistent intracellular bacterial and fungal infections and to granuloma formation.
Chronic Granulomatous Disease (CGD) has an estimated incidence of 1 in 200,000 to 1 in 250,000 live births worldwide. It affects males and females equally and is more common in certain ethnic groups, including those of Middle Eastern, North African, and Turkish descent.
The clinical presentation of CGD can vary widely depending on the specific genetic mutation and the degree of residual NADPH oxidase activity. However, the most common clinical manifestations include recurrent bacterial and fungal infections, particularly of the skin, lungs, and lymph nodes. These infections can be severe and life-threatening, especially if they are not diagnosed and treated promptly.
CGD patients are also at increased risk of developing granulomatous inflammation, which can affect various organs such as the liver, spleen, and gastrointestinal tract. The granulomas can cause organ dysfunction and may require surgical intervention in some cases.
In approximately two-thirds of patients, the first symptoms of CGD appear during the first year of life in the form of infections, dermatitis (sometimes seen at birth), gastrointestinal complications (obstruction or intermittent bloody diarrhea due to colitis), and a failure to thrive.
The clinical picture can be quite variable, with some infants having several of these complications and others appearing to be far less ill.
Cutaneous disease occurs in 60-70% of patients.
There is a poorly explained failure to switch off the inflammatory reaction with the formation of granuloma in many sites and poor wound healing.
CGD manifestations include recurrent infections; multiple granulomatous lesions of the lungs, liver, lymph nodes, and GI and GU tract; abscesses; lymphadenitis; hypergammaglobulinemia; elevated ESR; and anemia.

Adult case of X-linked Chronic Granulomatous Disease presenting with nasal and medial canthus skin ulceration, illustrating severe CGD-related granulomatous inflammation.

Common organ involvement in Chronic Granulomatous Disease, including lymph nodes, liver, colon, bone, lungs, stomach, urinary tract, and skin, reflecting the multisystem nature of CGD complications.
Diagnosis:
Nitroblue Tetrazolium Dye (NBT test):
The diagnosis of chronic granulomatous disease requires a combination of clinical suspicion, immunological testing, and genetic analysis. The gold standard for diagnosis is the demonstration of an inability of the neutrophils to reduce Nitroblue Tetrazolium Dye (NBT test), and the defect can be further defined by molecular techniques.

Normal NBT test: neutrophils showing dark formazan deposits, demonstrating preserved oxidative burst in the assessment of Chronic Granulomatous Disease.
The nitroblue tetrazolium (NBT) slide test is used for screening defects in NADPH oxidase. In this test, patient neutrophils are exposed to a stimulus, incubated with NBT, and made into a smear on a slide. Under the microscope, the number of neutrophils with dark granules of reaction product are counted. Normally, more than 95% of the granulocytes will be positive as shown above. In chronic granulomatous disease (CGD) there is an absent or reduced function of the respiratory burst, which is the intracellular process in neutrophils that is dependent upon the enzyme NADPH oxidase, which produces oxygen free radicals used to kill phagocytized organisms. The abnormal NBT test in CGD is shown below in which <5% of neutrophils stain.

Abnormal NBT test: neutrophils lacking formazan deposits, demonstrating a defective oxidative burst typical of Chronic Granulomatous Disease.
DHR Flow Cytometry Test:
The dihydrorhodamine (DHR) flow cytometry test is the preferred diagnostic assay nowadays for Chronic Granulomatous Disease because it provides a sensitive, quantitative assessment of neutrophil oxidative burst. Following stimulation, normal neutrophils convert non-fluorescent DHR into fluorescent rhodamine, whereas CGD neutrophils show absent or markedly reduced fluorescence. The DHR test reliably distinguishes affected patients, carriers of X-linked CGD, and individuals with residual NADPH oxidase activity, making it superior to the traditional NBT test for both diagnosis and monitoring.

DHR flow cytometry in Chronic Granulomatous Disease demonstrating three key patterns: normal oxidative burst (A), CGD carrier mosaic pattern (B), and absent oxidative burst in CGD (C).
Treatment:
The management of CGD involves a multidisciplinary approach that includes infectious disease specialists, immunologists, and hematologists. The primary goal is to prevent and treat infections promptly with appropriate antimicrobial agents.
Treatment is continuous prophylactic antibiotics, particularly trimethoprim/sulfamethoxazole (Septrin) 160/80 mg po bid. Oral antifungals are given as primary prophylaxis or are added if fungal infections occur even once; most useful are itraconazole po q 12 h (100 mg for patients < 13 yr; 200 mg for those ≥ 13 yr or weighing > 50 kg), voriconazole po q 12 h (100 mg for those weighing < 40 kg; 200 mg for those weighing ≥ 40 kg), or posaconazole (400 mg bid). Interferon-gamma may reduce the severity and frequency of infections and is usually included in the treatment regimen. The usual dose is 50 mcg/m2 sc 3 times/wk.
Granulocyte transfusions can be lifesaving when infections are severe. When preceded by pretransplantation chemotherapy, HLA-identical sibling bone marrow transplantation is usually successful.
Gene therapy is under study.
Prognosis:
The prognosis of Chronic Granulomatous Disease has improved substantially over recent decades due to earlier diagnosis, routine antimicrobial prophylaxis, interferon-γ therapy, and advances in supportive care. Survival into adulthood is now common, particularly in patients with milder genetic variants or residual NADPH oxidase activity. However, recurrent infections, inflammatory complications, granulomatous obstruction of the gastrointestinal or genitourinary tract, and hepatic or pulmonary involvement continue to impact long-term outcomes. Allogeneic hematopoietic stem cell transplantation offers the potential for cure and is associated with excellent survival in appropriately selected candidates, especially when performed early before irreversible organ damage occurs. Lifelong monitoring, rapid treatment of infections, and multidisciplinary management remain essential to improving quality of life and overall survival in individuals with Chronic Granulomatous Disease.
Summary:
Chronic Granulomatous Disease (CGD) is a rare inherited primary immunodeficiency caused by defects in the NADPH oxidase complex, leading to failure of phagocytes to produce reactive oxygen species (ROS) and destroy catalase-positive pathogens. As a result, patients experience recurrent life-threatening bacterial and fungal infections, granuloma formation, and inflammatory complications involving the lungs, skin, lymph nodes, gastrointestinal tract, and liver. CGD is most commonly X-linked but also occurs in autosomal recessive forms. Diagnosis relies on functional assays such as the dihydrorhodamine (DHR) test or NBT reduction test, supported by genetic analysis. Management includes antimicrobial prophylaxis, interferon-γ therapy, aggressive treatment of infections, and consideration of curative allogeneic stem cell transplantation. Early recognition and optimized multidisciplinary care significantly improve long-term outcomes in individuals with Chronic Granulomatous Disease.
Questions and Answers:
What is Chronic Granulomatous Disease?
Chronic Granulomatous Disease is a rare inherited immunodeficiency caused by defects in the NADPH oxidase complex, leading to impaired phagocyte oxidative burst and recurrent bacterial and fungal infections.
What causes Chronic Granulomatous Disease?
CGD is caused by mutations in genes encoding components of the NADPH oxidase enzyme complex, most commonly through X-linked inheritance but also via autosomal recessive variants.
What infections are common in Chronic Granulomatous Disease?
Patients with CGD are frequently affected by catalase-positive organisms such as Staphylococcus aureus, Serratia marcescens, Burkholderia cepacia, Nocardia, and Aspergillus species.
What organs are most affected in Chronic Granulomatous Disease?
CGD commonly involves the lungs, lymph nodes, skin, liver, gastrointestinal tract, bones, and urinary tract, often presenting with abscesses, granulomas, pneumonia, or colitis.
How is Chronic Granulomatous Disease diagnosed?
Diagnosis relies on functional neutrophil assays such as the DHR flow cytometry test or NBT test, followed by genetic testing to confirm the specific NADPH oxidase defect.
What does a normal NBT test look like in CGD assessment?
A normal NBT test shows neutrophils with dark formazan deposits, indicating intact oxidative burst and helping differentiate carriers and unaffected individuals from CGD patients.
What does an abnormal NBT test indicate?
An abnormal NBT test demonstrates absent or markedly reduced formazan staining, confirming defective neutrophil oxidative burst consistent with Chronic Granulomatous Disease.
What are the typical skin manifestations of Chronic Granulomatous Disease?
Common skin findings include recurrent abscesses, cellulitis, and granulomatous ulceration of areas such as the face, nasal bridge, and medial canthus.
What complications can occur in Chronic Granulomatous Disease?
CGD can lead to severe infections and granulomatous inflammation affecting organs such as the liver, colon, lungs, stomach, bones, and genitourinary tract.
How is Chronic Granulomatous Disease treated?
Management includes antimicrobial prophylaxis (trimethoprim-sulfamethoxazole and azoles), interferon-γ therapy, prompt treatment of infections, and consideration of curative stem cell transplantation.
Can Chronic Granulomatous Disease be cured?
The only curative option is allogeneic hematopoietic stem cell transplantation, which offers excellent outcomes when performed early in suitable candidates.
What is the life expectancy in Chronic Granulomatous Disease?
With modern prophylaxis and supportive care, many individuals with CGD survive well into adulthood, although prognosis varies depending on genetic subtype and severity of infections and inflammation.
Is Chronic Granulomatous Disease inherited?
Yes. Most cases are X-linked due to CYBB gene mutations, while others follow an autosomal recessive pattern affecting components of the NADPH oxidase system.
References:
Roos D, de Boer M. Molecular diagnosis of chronic granulomatous disease. Clin Exp Immunol. 2014;175(2):139–149.
Segal BH, Romani L, Puccetti P. Chronic granulomatous disease. Cell Mol Life Sci. 2009 Feb;66(4):553–558.
Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore). 2000;79(3):170–200.
Wiley Online Library. Chronic granulomatous disease case report. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/jdv.16291
Winkelstein JA, Marino MC, Johnston RB Jr, et al. Chronic granulomatous disease: report on a national registry of 368 patients. Medicine (Baltimore). 2000;79(3):155–169.
Marciano BE, Rosenzweig SD, Kleiner DE, et al. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. 2004;114(2):462–468.
Holland SM. Chronic granulomatous disease. Clin Rev Allergy Immunol. 2010;38(1):3–10.
Seger RA. Modern management of chronic granulomatous disease. Br J Haematol. 2008;140(3):255–266.
McLean-Tooke A, Aldridge C, Gilmour K, Higgins B, Hudson M, Spickett G. An unusual cause of granulomatous disease. BMC Clin Pathol. 2007;7:1. doi:10.1186/1472-6890-7-1.
Chronic Granulomatous Disease (CGD) — Symptoms and Sequelae. CGDPathways.com. Available from: https://www.cgdpathways.com/cgd-overview/symptoms-and-sequelae-of-chronic-granulomatous-disease
Keywords:
chronic granulomatous disease, CGD diagnosis, NADPH oxidase deficiency, phagocyte oxidative burst defect, DHR flow cytometry test, NBT test CGD, CGD symptoms, CGD treatment, CGD prognosis, CGD genetics, X-linked CGD, autosomal recessive CGD, CGD infections, CGD complications, granulomatous inflammation CGD, CGD carrier pattern, CGD skin lesions, CGD lung infections, CGD gastrointestinal granulomas, CGD liver abscess, CGD osteomyelitis, CGD lymphadenitis, CGD bone involvement, CGD immunodeficiency, CGD management, interferon gamma therapy CGD, CGD stem cell transplantation, CGD flow cytometry patterns, CGD neutrophil dysfunction
Related Posts:





Request Online Consultation With Dr M Abdou
Fee: US$100
Secure payment via PayPal (credit and debit cards accepted)
Pay Now