Gaucher’s Disease
Gaucher’s Disease is a rare genetic disorder that falls under the category of lysosomal storage diseases.
Gaucher’s Disease is an autosomal recessive condition most common in Jews.
Gaucher’s Disease is caused by mutations in the GBA gene, leading to deficient activity of the enzyme glucocerebrosidase. This deficiency results in the accumulation of glucocerebroside within macrophages, leading to the formation of Gaucher cells. These cells infiltrate various organs, primarily the liver, spleen, CNS, and bone marrow, causing a range of clinical manifestations.
The β-glucocerebrosidase deficiency causes deposition of glucocerebroside and related compounds, leading to hepatosplenomegaly, CNS changes and bony infiltration.
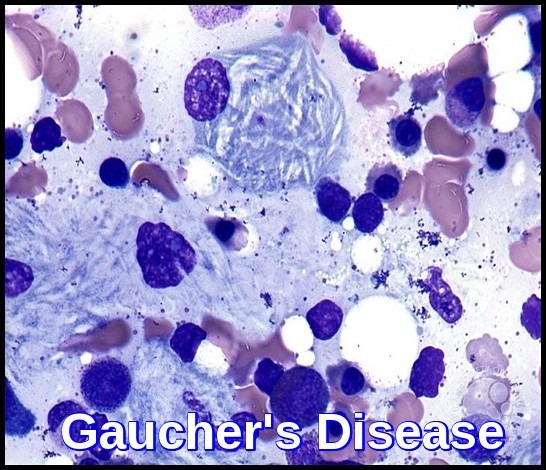
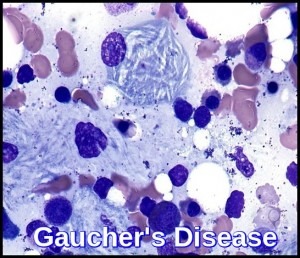
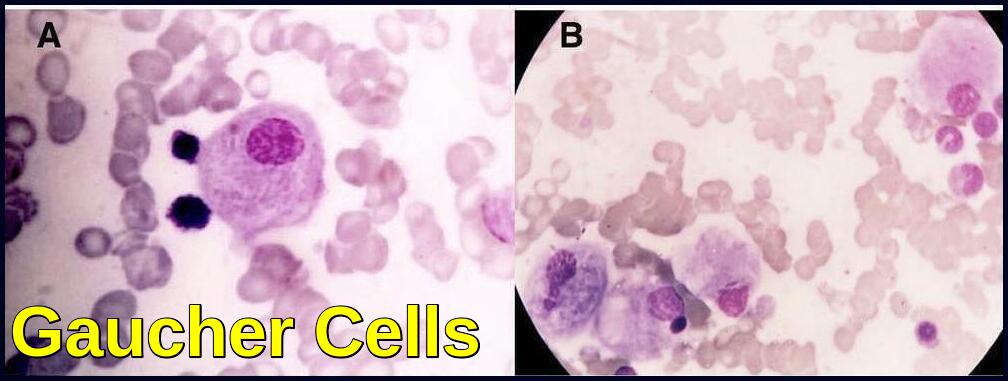
Glucocerebrosidase normally hydrolyzes glucocerebroside to glucose and ceramide. Genetic defects of the enzyme cause glucocerebroside accumulation in tissue macrophages through phagocytosis, forming Gaucher cells (with onion skin appearance). Accumulation of Gaucher cells in the perivascular spaces in the brain causes gliosis in the neuronopathic forms.
The course of the disease is very variable.
There are 3 types, which vary in epidemiology, enzyme activity, and manifestations:
Type I (non-neuronopathic) is the most common (90% of all patients). Residual enzyme activity is highest. Ashkenazi Jews are at greatest risk—patients with type 1 disease commonly present with painless splenomegaly, anemia, or thrombocytopenia.
Type II (acute neuronopathic) is rare, and residual enzyme activity in this type is lowest. Onset occurs during infancy. Symptoms and signs are progressive neurologic deterioration (eg, rigidity, seizures) and death by age 2 years.
Type III (subacute neuronopathic) falls between types I and II in incidence, enzyme activity, and clinical severity.
The blood film often shows features of hypersplenism and typical Gaucher cells are found in the bone marrow.
Gaucher Cells in BM Aspirate
Clinical Manifestations:
The clinical presentation of Gaucher’s Disease varies widely, ranging from asymptomatic to severe systemic involvement. Common symptoms include hepatosplenomegaly, anemia, thrombocytopenia, and bone abnormalities such as osteopenia and avascular necrosis. Additionally, patients may experience fatigue, easy bruising, and a predisposition to fractures.
Diagnosis:
Accurate and timely diagnosis of Gaucher’s Disease is essential for initiating appropriate management. Hematologists often utilize a combination of clinical findings, imaging studies, and laboratory tests. The demonstration of Gaucher cells in bone marrow aspirates or biopsies is a definitive diagnostic method. Molecular genetic testing to identify GBA gene mutations further supports the diagnosis.
The diagnosis can be confirmed through measurement of glucocerebrosidase activity in WBCs. A finding of less than 15% of mean normal activity is diagnostic; carriers are detected, and types are distinguished by mutation analysis.
Case Report:
A 6-year-old boy presented with splenomegaly 15 cm below the left costal margin with a history of anemia since the age of 8 months. The liver was palpable to approximately 1 cm below the right costal margin. Abdominal ultrasound confirmed marked splenomegaly and hepatomegaly. Complete blood counts revealed pancytopenia. WBC count was: 2.7 x 10^9/L, Hemoglobin was : 10.0 g/dL, PLT: 98 x 10^9/L. A bone marrow biopsy was performed due to the patient’s persistent pancytopenia and splenomegaly. The bone marrow aspirate showed normal erythroid and myeloid maturation. Megakaryocytes were present in normal numbers. Individual as well as large clusters and aggregates of macrophages with eccentric nuclei, abundant blue-gray cytoplasm and wrinkled tissue paper appearance are seen.
Enzymatic quantification of β-glucocerebrosidase was performed which confirmed it’s deficit.
Gaucher’s Disease ( GD) is usually diagnosed by the demonstration of characteristic “Gaucher cells” in the bone marrow. Pseudo-Gaucher cells have occasionally been described in various hematologic malignancies including multiple myeloma, myelodysplastic syndrome, lymphomas, chronic myelogenous leukemia and thalassemia.
Therefore, detection of reduced or absent β-glucosidase (glucocerebrosidase) enzyme activity is the gold standard for the diagnosis of all the variants of GD.
Treatment:
Enzyme replacement therapy (ERT) has revolutionized the management of Gaucher’s Disease. Recombinant glucocerebrosidase is administered intravenously, effectively reducing the accumulation of glucocerebroside and ameliorating symptoms. Substrate reduction therapy (SRT) and, in severe cases, hematopoietic stem cell transplantation (HSCT) may also be considered.
Treatment for types I and III include enzyme replacement with glucocerebrosidase, and sometimes miglustat, eliglustat, splenectomy, or stem cell or bone marrow transplantation; there is no treatment for type II.
The 3 FDA-approved drugs available for treating Gaucher disease include:
Cerezyme® (imiglucerase)
ELELYSO® (taliglucerase alfa)
VPRIV® (velaglucerase alfa)
N.B.-specific enzyme replacement therapy is very expensive!
References:
Mistry PK, Taddei T, vom Dahl S, Rosenbloom BE. Gaucher disease and malignancy: a model for cancer pathogenesis in an inborn error of metabolism. Crit Rev Oncog. 2013;18(3):235-246.
Zimran A, Belmatoug N, Bembi B, et al. Demographics and patient characteristics of 1209 patients with Gaucher disease: Descriptive analysis from the Gaucher Outcome Survey (GOS). Am J Hematol. 2018;93(2):205-212.
Dr Rupali Parikh, Bhatia Hospital, Mumbai. Gaucher’s cells in bone marrow aspirate smears | ASH Image Bank | American Society of Hematology https://imagebank.hematology.org/image/62962/gauchers-cells-in-bone-marrow-aspirate-smears